Factor XII
Coagulation factor XII, also known as Hageman factor, is a plasma protein. It is the zymogen form of factor XIIa, an enzyme (EC 3.4.21.38) of the serine protease (or serine endopeptidase) class. In humans, factor XII is encoded by the F12 gene.[5]





Structure
Human Factor XII is 596 amino acids long and consists of two chains, the heavy chain (353 residues) and light chain (243 residues) held together by a disulfide bond. It is 80,000 daltons. Its heavy chain contains two fibronectin-type domains (type I and II), two epidermal growth factor-like domains, a kringle domain, and a proline-rich region, and its light chain contains the protease domain. The structure of the FnI-EGF-like tandem domain of coagulation factor XII has been solved by X-ray crystallography.[6][7] Crystal structures of the FXII light chain has also been determined unbound (β-FXII) and bound (β-FXIIa) to inhibitors.[8][9][10]
Function
Factor XII is part of the coagulation cascade and activates factor XI and prekallikrein in vitro. Factor XII itself is activated to factor XIIa by negatively charged surfaces, such as glass. This is the starting point of the intrinsic pathway.[11] Factor XII can also be used to start coagulation cascades in laboratory studies.[12]
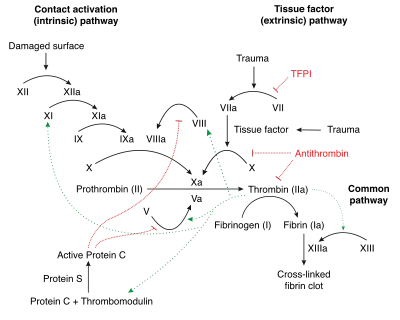
In vivo, factor XII is activated by contact to polyanions. Activated platelets secrete inorganic polymers, polyphosphates. Contact to polyphosphates activates factor XII and initiates fibrin formation by the intrinsic pathway of coagulation with critical importance for thrombus formation. Targeting polyphosphates with phosphatases interfered with procoagulant activity of activated platelets and blocked platelet-induced thrombosis in mice. Addition of polyphosphates restored defective plasma clotting of Hermansky–Pudlak syndrome patients, indicating that the inorganic polymer is the endogenous factor XII activator in vivo. Platelet polyphosphate-driven factor XII activation provides the link from primary hemostasis (formation of a platelet plug) to secondary hemostasis (fibrin meshwork formation).[13]
Genetics
The gene for factor XII is located on the tip of the long arm of the fifth chromosome (5q33-qter).[5]
Role in disease
Factor XII deficiency is a rare disorder that is inherited in an autosomal recessive manner.[14] Unlike other clotting factor deficiencies, factor XII deficiency is totally asymptomatic and does not cause excess bleeding.[14] Mice lacking the gene for factor XII, however, are less susceptible to thrombosis. The protein seems to be involved in the later stages of clot formation rather than the first occlusion of damages in the blood vessel wall.[15]
Factor XII does play an important role in clot formation during in vitro measurements of the partial thromboplastin time, which causes these measurements to be markedly prolonged in patients with factor XII deficiency, usually well beyond even what is seen in hemophilia A, hemophilia B, or factor XI deficiency.[14] As a result, the main concern related to factor XII deficiency is the unnecessary testing, delay in care, worry, etc. that may be prompted by the abnormal lab result.[14] All of this, including the mechanism of inheritance, also holds true for the other contact factors, prekallikrein (Fletcher factor) and high molecular weight kininogen.[14]
Excess levels of factor XII can predispose individuals towards greater risk of venous thrombosis due to factor XII's role as one of the catalysts for conversion of plasminogen to its active fibrinolytic form of plasmin.[16]
Factor XII is also activated by endotoxins, especially lipid A. Factor XII is hageman factor
History
Hageman factor was first discovered in 1955 when a routine preoperative blood sample of the 37-year-old railroad brakeman John Hageman (1918) was found to have prolonged clotting time in test tubes, even though he had no hemorrhagic symptoms. Hageman was then examined by hematologist Oscar Ratnoff, who found that Hageman lacked a previously unidentified clotting factor.[17] Ratnoff later found that the Hageman factor deficiency is an autosomal recessive disorder, after examining several related people who had the deficiency. Paradoxically, pulmonary embolism contributed to Hageman's death after an occupational accident in 1968. Since then, case studies and clinical studies identified an association between thrombosis and Factor XII deficiency. Hepatocytes express blood coagulation factor XII.[18]
References
- GRCh38: Ensembl release 89: ENSG00000131187 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000021492 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Cool DE, MacGillivray RT (October 1987). "Characterization of the human blood coagulation factor XII gene. Intron/exon gene organization and analysis of the 5'-flanking region". The Journal of Biological Chemistry. 262 (28): 13662–73. PMID 2888762.
- Stavrou E, Schmaier AH (March 2010). "Factor XII: what does it contribute to our understanding of the physiology and pathophysiology of hemostasis & thrombosis". Thrombosis Research. 125 (3): 210–5. doi:10.1016/j.thromres.2009.11.028. PMC 2851158. PMID 20022081.
- Beringer DX, Kroon-Batenburg LM (February 2013). "The structure of the FnI-EGF-like tandem domain of coagulation factor XII solved using SIRAS". Acta Crystallographica Section F. 69 (Pt 2): 94–102. doi:10.1107/S1744309113000286. PMC 3564606. PMID 23385745.
- Dementiev A, Silva A, Yee C, Li Z, Flavin MT, Sham H, Partridge JR (March 2018). "Structures of human plasma β-factor XIIa cocrystallized with potent inhibitors". Blood Advances. 2 (5): 549–558. doi:10.1182/bloodadvances.2018016337. PMC 5851424. PMID 29519898.
- Pathak M, Wilmann P, Awford J, Li C, Hamad BK, Fischer PM, et al. (April 2015). "Coagulation factor XII protease domain crystal structure". Journal of Thrombosis and Haemostasis. 13 (4): 580–91. doi:10.1111/jth.12849. PMC 4418343. PMID 25604127.
- Pathak M, Manna R, Li C, Kaira BG, Hamad BK, Belviso BD, et al. (June 2019). "Crystal structures of the recombinant β-factor XIIa protease with bound Thr-Arg and Pro-Arg substrate mimetics". Acta Crystallographica Section D. 75 (Pt 6): 578–591. doi:10.1107/S2059798319006910. PMID 31205020.
- Guyton AC, Hall JE. Textbook of Medical Physiology (PDF) (11th ed.). pp. 462–463. ISBN 0-7216-0240-1.
- Renné T, Schmaier AH, Nickel KF, Blombäck M, Maas C (November 2012). "In vivo roles of factor XII". Blood. 120 (22): 4296–303. doi:10.1182/blood-2012-07-292094. PMC 3507141. PMID 22993391.
- Müller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, et al. (December 2009). "Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo". Cell. 139 (6): 1143–56. doi:10.1016/j.cell.2009.11.001. PMC 2796262. PMID 20005807.
- Wagenman BL, Townsend KT, Mathew P, Crookston KP (June 2009). "The laboratory approach to inherited and acquired coagulation factor deficiencies". Clinics in Laboratory Medicine. 29 (2): 229–52. doi:10.1016/j.cll.2009.04.002. PMID 19665676.
- Renné T, Pozgajová M, Grüner S, Schuh K, Pauer HU, Burfeind P, et al. (July 2005). "Defective thrombus formation in mice lacking coagulation factor XII". The Journal of Experimental Medicine. 202 (2): 271–81. doi:10.1084/jem.20050664. PMC 2213000. PMID 16009717.
- Kroll, Michael H. (2001). Manual of Coagulation Disorders. Blackwell Science. pp. 3–4, 206–207. ISBN 0-86542-446-2.
- Ratnoff OD, Margolius A (1955). "Hageman trait: an asymptomatic disorder of blood coagulation". Transactions of the Association of American Physicians. 68: 149–54. PMID 13299324.
- Gordon EM, Gallagher CA, Johnson TR, Blossey BK, Ilan J (April 1990). "Hepatocytes express blood coagulation factor XII (Hageman factor)". The Journal of Laboratory and Clinical Medicine. 115 (4): 463–9. PMID 2324612.
Further reading
- Girolami A, Randi ML, Gavasso S, Lombardi AM, Spiezia F (April 2004). "The occasional venous thromboses seen in patients with severe (homozygous) FXII deficiency are probably due to associated risk factors: a study of prevalence in 21 patients and review of the literature". Journal of Thrombosis and Thrombolysis. 17 (2): 139–43. doi:10.1023/B:THRO.0000037670.42776.cd. PMID 15306750. S2CID 19336633.
- Renné T, Gailani D (July 2007). "Role of Factor XII in hemostasis and thrombosis: clinical implications". Expert Review of Cardiovascular Therapy. 5 (4): 733–41. doi:10.1586/14779072.5.4.733. PMID 17605651. S2CID 41250936.
- Harris RJ, Ling VT, Spellman MW (March 1992). "O-linked fucose is present in the first epidermal growth factor domain of factor XII but not protein C". The Journal of Biological Chemistry. 267 (8): 5102–7. PMID 1544894.
- McMullen BA, Fujikawa K, Davie EW (February 1991). "Location of the disulfide bonds in human plasma prekallikrein: the presence of four novel apple domains in the amino-terminal portion of the molecule". Biochemistry. 30 (8): 2050–6. doi:10.1021/bi00222a007. PMID 1998666.
- Miyata T, Kawabata S, Iwanaga S, Takahashi I, Alving B, Saito H (November 1989). "Coagulation factor XII (Hageman factor) Washington D.C.: inactive factor XIIa results from Cys-571----Ser substitution". Proceedings of the National Academy of Sciences of the United States of America. 86 (21): 8319–22. Bibcode:1989PNAS...86.8319M. doi:10.1073/pnas.86.21.8319. PMC 298272. PMID 2510163.
- Bernardi F, Marchetti G, Patracchini P, del Senno L, Tripodi M, Fantoni A, et al. (May 1987). "Factor XII gene alteration in Hageman trait detected by TaqI restriction enzyme". Blood. 69 (5): 1421–4. doi:10.1182/blood.V69.5.1421.1421. PMID 2882793.
- Cool DE, MacGillivray RT (October 1987). "Characterization of the human blood coagulation factor XII gene. Intron/exon gene organization and analysis of the 5'-flanking region". The Journal of Biological Chemistry. 262 (28): 13662–73. PMID 2888762.
- Que BG, Davie EW (April 1986). "Characterization of a cDNA coding for human factor XII (Hageman factor)". Biochemistry. 25 (7): 1525–8. doi:10.1021/bi00355a009. PMID 3011063.
- Royle NJ, Nigli M, Cool D, MacGillivray RT, Hamerton JL (March 1988). "Structural gene encoding human factor XII is located at 5q33-qter". Somatic Cell and Molecular Genetics. 14 (2): 217–21. doi:10.1007/BF01534407. PMID 3162339. S2CID 7868690.
- Citarella F, Tripodi M, Fantoni A, Bernardi F, Romeo G, Rocchi M (December 1988). "Assignment of human coagulation factor XII (fXII) to chromosome 5 by cDNA hybridization to DNA from somatic cell hybrids". Human Genetics. 80 (4): 397–8. doi:10.1007/BF00273661. PMID 3198120. S2CID 13428786.
- Henry ML, Everson B, Ratnoff OD (May 1988). "Inhibition of the activation of Hageman factor (factor XII) by beta 2-glycoprotein I". The Journal of Laboratory and Clinical Medicine. 111 (5): 519–23. PMID 3361230.
- Chung DW, Fujikawa K, McMullen BA, Davie EW (May 1986). "Human plasma prekallikrein, a zymogen to a serine protease that contains four tandem repeats". Biochemistry. 25 (9): 2410–7. doi:10.1021/bi00357a017. PMID 3521732.
- Tripodi M, Citarella F, Guida S, Galeffi P, Fantoni A, Cortese R (April 1986). "cDNA sequence coding for human coagulation factor XII (Hageman)". Nucleic Acids Research. 14 (7): 3146. doi:10.1093/nar/14.7.3146. PMC 339730. PMID 3754331.
- Cool DE, Edgell CJ, Louie GV, Zoller MJ, Brayer GD, MacGillivray RT (November 1985). "Characterization of human blood coagulation factor XII cDNA. Prediction of the primary structure of factor XII and the tertiary structure of beta-factor XIIa". The Journal of Biological Chemistry. 260 (25): 13666–76. PMID 3877053.
- McMullen BA, Fujikawa K (May 1985). "Amino acid sequence of the heavy chain of human alpha-factor XIIa (activated Hageman factor)". The Journal of Biological Chemistry. 260 (9): 5328–41. PMID 3886654.
- de Grouchy J, Turleau C (1975). "Tentative localization of a Hageman (Factor XII) locus on 7q, probably the 7q35 band". Humangenetik. 24 (3): 197–200. doi:10.1007/bf00283584. PMID 4140832. S2CID 11075182.
- Fujikawa K, McMullen BA (September 1983). "Amino acid sequence of human beta-factor XIIa". The Journal of Biological Chemistry. 258 (18): 10924–33. PMID 6604055.
- Hovinga JK, Schaller J, Stricker H, Wuillemin WA, Furlan M, Lämmle B (August 1994). "Coagulation factor XII Locarno: the functional defect is caused by the amino acid substitution Arg 353-->Pro leading to loss of a kallikrein cleavage site". Blood. 84 (4): 1173–81. doi:10.1182/blood.V84.4.1173.1173. PMID 8049433.
- Schloesser M, Hofferbert S, Bartz U, Lutze G, Lämmle B, Engel W (July 1995). "The novel acceptor splice site mutation 11396(G-->A) in the factor XII gene causes a truncated transcript in cross-reacting material negative patients" (PDF). Human Molecular Genetics. 4 (7): 1235–7. doi:10.1093/hmg/4.7.1235. PMID 8528215.
- Hofferbert S, Müller J, Köstering H, von Ohlen WD, Schloesser M (June 1996). "A novel 5'-upstream mutation in the factor XII gene is associated with a TaqI restriction site in an Alu repeat in factor XII-deficient patients". Human Genetics. 97 (6): 838–41. doi:10.1007/BF02346200. PMID 8641707. S2CID 9589165.
External links
- The MEROPS online database for peptidases and their inhibitors: S01.211
- Factor+XII at the US National Library of Medicine Medical Subject Headings (MeSH)
- Overview of all the structural information available in the PDB for UniProt: P00748 (Coagulation factor XII) at the PDBe-KB.