Apparent mineralocorticoid excess syndrome
Apparent mineralocorticoid excess is an autosomal recessive[1] disorder causing hypertension (high blood pressure) and hypokalemia (abnormally low levels of potassium). It results from mutations in the HSD11B2 gene, which encodes the kidney isozyme of 11β-hydroxysteroid dehydrogenase type 2. In an unaffected individual, this isozyme inactivates circulating cortisol to the less active metabolite cortisone. The inactivating mutation leads to elevated local concentrations of cortisol in the aldosterone sensitive tissues like the kidney. Cortisol at high concentrations can cross-react and activate the mineralocorticoid receptor due to the non-selectivity of the receptor, leading to aldosterone-like effects in the kidney. This is what causes the hypokalemia, hypertension, and hypernatremia associated with the syndrome. Patients often present with severe hypertension and end-organ changes associated with it like left ventricular hypertrophy, retinal, renal and neurological vascular changes along with growth retardation and failure to thrive. In serum both aldosterone and renin levels are low.[2]
| Apparent mineralocorticoid excess syndrome | |
|---|---|
| Other names | AME |
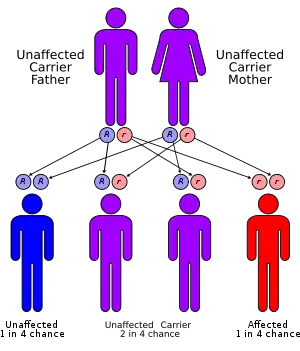 | |
| Apparent mineralocorticoid excess syndrome has an autosomal recessive pattern of inheritance | |
| Specialty | Medical genetics, endocrinology |
Signs and symptoms
This disorder presents similarly to hyperaldosteronism, leading to feedback inhibition of aldosterone. Common symptoms include hypertension, hypokalemia, metabolic alkalosis, and low plasma renin activity.
DOC excess syndrome is an excessive secretion of 21-hydroxyprogesterone also called 11-Deoxycorticosterone from adrenal glands and may cause mineralocorticoid hypertension.[3][4][5]
Genetics
AME is inherited in an autosomal recessive manner.[1] This means the defective gene responsible for the disorder is located on an autosome, and two copies of the defective gene (one inherited from each parent) are required in order to be born with the disorder. The parents of an individual with an autosomal recessive disorder both carry one copy of the defective gene, but usually do not experience any signs or symptoms of the disorder.
Diagnosis
Other conditions such as Liddle's Syndrome can mimic the clinical features of AME, so diagnosis can be made by calculating the ratio of free urinary cortisol to free urinary cortisone. Since AME patients create less cortisone, the ratio will much be higher than non-affected patients.[6] Alternatively, one could differentiate between the two syndromes by administering a potassium-sparing diuretic. Patients with Liddle's syndrome will only respond to a diuretic that binds the ENaC channel, whereas those with AME will respond to a diuretic that binds to ENaC or the mineralcorticoid receptor.
Treatment
The treatment for AME is based on the blood pressure control with Aldosterone antagonist like Spironolactone which also reverses the hypokalemic metabolic alkalosis and other anti-hypertensives. Renal transplant is found curative in almost all clinical cases.AME is exceedingly rare, with fewer than 100 cases recorded worldwide.[6]
Liquorice consumption may also cause a temporary form of AME due to its ability to block 11β-hydroxysteroid dehydrogenase type 2, in turn causing increased levels of cortisol.[7][8] Cessation of licorice consumption will reverse this form of AME.
See also
- Inborn errors of steroid metabolism
- 11β-Hydroxylase I deficiency
- Hyperaldosteronism
- Pseudohyperaldosteronism
- Glucocorticoid-remediable aldosteronism
- Aldosterone and aldosterone synthase
- Maria New
References
- Levtchenko, E. N.; Deinum, J.; Knoers, N. V. a. M.; Hermus, A. R.; Monnens, L. a. H.; Lenders, J. W. M. (2007). "Van gen naar ziekte; 'apparent mineralocorticoid excess'-syndroom, een syndroom met ogenschijnlijke overmaat aan mineralocorticoïden" [From gene to disease; 'apparent mineralocorticoid excess' syndrome, a syndrome with an apparent excess of mineral corticoids]. Nederlands Tijdschrift voor Geneeskunde (in Dutch). 151 (12): 692–694. hdl:11370/df94307a-2281-4450-93bf-a30c7bc2d0ab. PMID 17447595.
- Al-Harbi, Taiba; Al-Shaikh, Adnan (1 January 2012). "Apparent mineralocorticoid excess syndrome: report of one family with three affected children". Journal of Pediatric Endocrinology and Metabolism. 25 (11–12): 1083–8. doi:10.1515/jpem-2012-0113. PMID 23329753. S2CID 21281031.
- Marques, Pedro; Tufton, Nicola; Bhattacharya, Satya; Caulfield, Mark; Akker, Scott A (3 May 2019). "Hypertension due to a deoxycorticosterone-secreting adrenal tumour diagnosed during pregnancy". Endocrinology, Diabetes & Metabolism Case Reports. 2019. doi:10.1530/EDM-18-0164. PMC 6499913. PMID 31051469.
- Gupta, Saurabh; Melendez, Jose; Khanna, Apurv (2010). "Deoxycorticosterone Producing Tumor as a Cause of Resistant Hypertension". Case Reports in Medicine. 2010: 372719. doi:10.1155/2010/372719. PMC 2909735. PMID 20671982.
- Deoxycorticosterone (DOC) at eMedicine
- Palermo M, Quinkler M, Stewart PM (Oct 2004). "Apparent mineralocorticoid excess syndrome: an overview" (PDF). Arq Bras Endocrinol Metabol. 48 (5): 687–696. doi:10.1590/S0004-27302004000500015. PMID 15761540.
- "HSD11B2 Gene". GeneCards.
- Sontia, Bruno; Mooney, Jan; Gaudet, Lise; Touyz, Rhian M. (February 2008). "Pseudohyperaldosteronism, Liquorice, and Hypertension". The Journal of Clinical Hypertension. 10 (2): 153–157. doi:10.1111/j.1751-7176.2008.07470.x. PMID 18256580. S2CID 20098685.
External links
- Apparent mineralocorticoid excess at NIH's Office of Rare Diseases
| Classification | |
|---|---|
| External resources |