Very long-chain acyl-coenzyme A dehydrogenase deficiency
Very long-chain acyl-coenzyme A dehydrogenase deficiency is a fatty-acid metabolism disorder which prevents the body from converting certain fats to energy, particularly during periods without food.[1][2][3]
| Very long-chain acyl-coenzyme A dehydrogenase deficiency | |
|---|---|
| Other names | VLCADD |
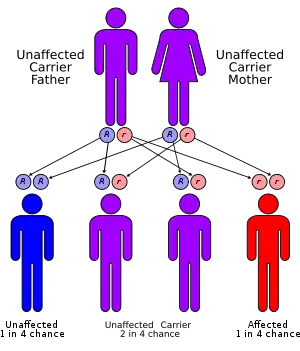 | |
| Very long-chain acyl-coenzyme: A dehydrogenase deficiency has an autosomal recessive pattern of inheritance. | |
Those affected by this disorder have inadequate levels of an enzyme that breaks down a group of fats called very long-chain fatty acids.[4]
Signs and symptoms
Signs and symptoms can include:[5][6]
- hypoglycemia
- lethargy
- hepatomegaly
- muscle pain
- cardiomyopathy
- Early onset-pericardial effusion
- heart arrhythmias
- vomiting
- *coma
- death
- Rhabdomyolysis
- Hypoketotic Hypoglycemia
Causes
VLCAD (very long-chain-acyl-dehydrogenase) deficiency is exclusively linked to genetic mutations in DNA. A change of the gene that codes for very long-chain-acyl-CoA-dehydrogenase (VLCAD) results in a deficiency or malfunction of the produced VLCAD enzyme.[7] This mutation occurs on chromosome 17 and can be altered via a variety of pathways.[4] These can range from frameshift mutations, deletion mutations, insertion mutations, and missense mutations. All of which cause the enzyme to function differently in the mitochondria, or in some cases not at all.[4] Due to this mutation, effective levels of very long-chain-acyl-CoA-dehydrogenase are low or absent in the body, giving rise to the array of symptoms listed above.[4][7]
Genetics
Mutations in the ACADVL gene lead to inadequate levels of an enzyme called very long-chain acyl-coenzyme A (CoA) dehydrogenase. Without this enzyme, long-chain fatty acids from food and fats stored in the body cannot be degraded and processed. As a result, these fatty acids are not converted into energy, which can lead to characteristic signs and symptoms of this disorder, such as lethargy and hypoglycemia. Levels of very long-chain fatty acids or partially degraded fatty acids may build up in tissues and can damage the heart, liver, and muscles, causing more serious complications.
VLCAD deficiency is characterized as an inherited genetic disorder. The mutations that occur within the gene itself are recessive, meaning that an individual has to acquire both recessive mutated genes in order for the disease to manifest.[4] There are various forms of the disease that can be manifested in infancy, adolescence, and adulthood.[8] However, it is still unknown at to what causes the disease to manifest itself in the different life stages.
Diagnosis
Typically, initial signs and symptoms of this disorder occur during infancy and include low blood sugar (hypoglycemia), lack of energy (lethargy), and muscle weakness. There is also a high risk of complications such as liver abnormalities and life-threatening heart problems. Symptoms that begin later in childhood, adolescence, or adulthood tend to be milder and usually do not involve heart problems. Episodes of very long-chain acyl-coenzyme A dehydrogenase deficiency can be triggered by periods of fasting, illness, and exercise.
It is common for babies and children with the early and childhood types of VLCAD to have episodes of illness known as metabolic crises. Some of the first symptoms of a metabolic crisis are: extreme sleepiness, behavior changes, irritable mood, poor appetite. Some of these other symptoms of VLCAD in infants may also follow: fever, nausea, diarrhea, vomiting, hypoglycemia. Evaluation of symptom combinations can aid in a positive diagnosis of VLCAD.[9] Since symptoms vary depending on age and onset of the patient, consultation with a metabolic specialist should be considered. Diagnosis is further confirmed through genetic analysis of the VLCAD gene.[9]
Treatment
Treatment and management of VLCAD deficiency involve dietary restrictions as well as implementation of proper hydration to avoid further complications. Hospitalization due to VLCAD deficiency can be treated with intravenous (IV) glucose for hydration and alkalization of urine and prevention of renal malfunction or failure.[10] Avoidance of fasting periods, high-fat diets, and dehydration is recommended for those who are affected. A diet consisting of low-fat intake and supplemental calories is common for management of VLCAD deficiency. If a metabolic crisis is not treated, a child with VLCAD can develop: breathing problems, seizures, coma, sometimes leading to death.
Prognosis
Medical screening can confirm occurrences of VLCAD most often in neonatal and infancy stages. Approximately half of all patients show signs of VLCAD deficiency during the neonatal period, one-fourth present later in the first year of infancy, and the final quarter is split between manifestations in childhood and adulthood. Comorbidity of cardiomyopathy, arrhythmias[3] and rhabdomyolysis are extremely common in patients under 1 year old which can lead to complications later in life. Loss of awareness or seizure can occur from hypoketotic hypoglycemia,[3] which is often fatal if not caught in screening. However, prompt treatment shows high promise for improvement. Late-onset myopathic sufferers may only experience muscle-related, vague, sporadic symptoms, and may never be diagnosed.[3] There is an extremely high genotype-phenotype correlation in a presentation. Mitigation of VLCAD symptoms can be achieved through dietary management.
References
- Leslie, Nancy D.; Valencia, C. Alexander; Strauss, Arnold W.; Connor, Jessica A.; Zhang, Kejian (1993-01-01). "Very Long-Chain Acyl-Coenzyme a Dehydrogenase Deficiency". In Pagon, Roberta A.; Adam, Margaret P.; Ardinger, Holly H.; Wallace, Stephanie E.; Amemiya, Anne; Bean, Lora J.H.; Bird, Thomas D.; Ledbetter, Nikki; Mefford, Heather C. (eds.). GeneReviews. Seattle (WA): University of Washington, Seattle. PMID 20301763.update 2014
- Reference, Genetics Home. "VLCAD deficiency". Genetics Home Reference. Retrieved 2017-02-27.
- "VLCAD deficiency | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2018-04-17.
- Reference, Genetics Home. "VLCAD deficiency". Genetics Home Reference. Retrieved 2018-04-17.
- "Very Long Chain Acyl CoA Dehydrogenase Deficiency (LCAD)".
- https://rarediseases.info.nih.gov/diseases/5508/vlcad-deficiency
- "VLCAD deficiency | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2019-11-13.
- Very Long-Chain Acyl-CoA Dehydrogenase (VLCAD) Deficiency Information for Healthcare Professionals" (PDF). Kansas Department of Health and Environment. 8/13/2014. Retrieved October 16, 2019
- "American College of Medical Genetics ACT Sheet" (PDF). American College of Medical Genetics. 2010. Retrieved October 8, 2019.
- Leslie, N. D.; Valencia, C. A.; Strauss, A. W.; Zhang, K.; Adam, M. P.; Ardinger, H. H.; Pagon, R. A.; Wallace, S. E.; Bean LJH; Stephens, K.; Amemiya, A. (1993). "Very Long-Chain Acyl-Coenzyme A Dehydrogenase Deficiency". PMID 20301763. Cite journal requires
|journal=(help)
External links
| Classification |
|---|