Tumour heterogeneity
Tumour heterogeneity describes the observation that different tumour cells can show distinct morphological and phenotypic profiles, including cellular morphology, gene expression, metabolism, motility, proliferation, and metastatic potential.[1] This phenomenon occurs both between tumours (inter-tumour heterogeneity) and within tumours (intra-tumour heterogeneity). A minimal level of intra-tumour heterogeneity is a simple consequence of the imperfection of DNA replication: whenever a cell (normal or cancerous) divides, a few mutations are acquired[2]—leading to a diverse population of cancer cells.[3] The heterogeneity of cancer cells introduces significant challenges in designing effective treatment strategies. However, research into understanding and characterizing heterogeneity can allow for a better understanding of the causes and progression of disease. In turn, this has the potential to guide the creation of more refined treatment strategies that incorporate knowledge of heterogeneity to yield higher efficacy.[4]
Tumour heterogeneity has been observed in leukemias,[5] breast,[6] prostate,[7][8][9] colon,[10][11][12] brain,[13] esophagus,[14] head and neck,[15] bladder[16] and gynecological carcinomas,[17] liposarcoma,[18] and multiple myeloma.[19]
Models of heterogeneity
There are two models used to explain the heterogeneity of tumour cells. These are the cancer stem cell model and the clonal evolution model. The models are not mutually exclusive, and it is believed that they both contribute to heterogeneity in varying amounts across different tumour types.[20]
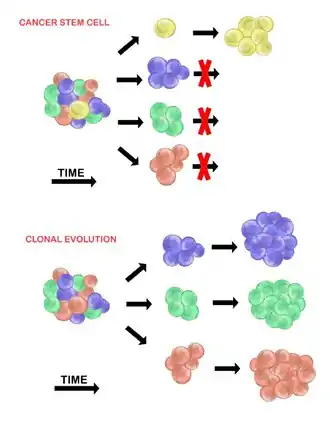
Cancer stem cells
The cancer stem cell model asserts that within a population of tumour cells, there is only a small subset of cells that are tumourigenic (able to form tumours). These cells are termed cancer stem cells (CSCs), and are marked by the ability to both self-renew and differentiate into non-tumourigenic progeny. The CSC model posits that the heterogeneity observed between tumour cells is the result of differences in the stem cells from which they originated. Stem cell variability is often caused by epigenetic changes, but can also result from clonal evolution of the CSC population where advantageous genetic mutations can accumulate in CSCs and their progeny (see below).[20]
Evidence of the cancer stem cell model has been demonstrated in multiple tumour types including leukemias,[21][22] glioblastoma,[23] breast cancer,[24] and prostate cancer.[25]
However, the existence of CSCs is still under debate. One reason for this is that markers for CSCs have been difficult to reproduce across multiple tumours. Further, methods for determining tumourigenic potential utilize xenograft models. These methods suffer from inherent limitations such as the need to control immune response in the transplant animal, and the significant difference in environmental conditions from the primary tumour site to the xenograft site (e.g. absence of required exogenous molecules or cofactors).[26] This has caused some doubt about the accuracy of CSC results and the conclusions about which cells have tumourigenic potential.
Clonal evolution
The clonal evolution model was first proposed in 1976 by Peter Nowell.[27] In this model, tumours arise from a single mutated cell, accumulating additional mutations as it progresses. These changes give rise to additional subpopulations, and each of these subpopulations has the ability to divide and mutate further. This heterogeneity may give rise to subclones that possess an evolutionary advantage over the others within the tumour environment, and these subclones may become dominant in the tumour over time.[28][29] When proposed, this model allowed for the understanding of tumour growth, treatment failure, and tumour aggression that occurs during the natural process of tumour formation.[28]
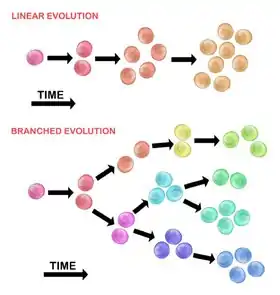
Evolution of the initial tumour cell may occur by two methods:
Linear expansion
Sequentially ordered mutations accumulate in driver genes, tumour suppressor genes, and DNA repair enzymes, resulting in clonal expansion of tumour cells. Linear expansion is less likely to reflect the endpoint of a malignant tumour[30] because the accumulation of mutations is stochastic in heterogeneic tumours.
Branched expansion
Expansion into multiple subclonal populations occurs through a splitting mechanism.[28] This method is more associated with tumour heterogeneity than linear expansion. The acquisition of mutations is random as a result of increased genomic instability with each successive generation. The long-term mutational accumulation may provide a selective advantage during certain stages of tumour progression. The tumor microenvironment may also contribute to tumour expansion, as it is capable of altering the selective pressures that the tumour cells are exposed to.[30]
Types and causes of heterogeneity
Multiple types of heterogeneity have been observed between tumour cells, stemming from both genetic and non-genetic variability.[31]
Genetic heterogeneity
Genetic heterogeneity is a common feature of tumour genomes, and can arise from multiple sources. Some cancers are initiated when exogenous factors introduce mutations, such as ultraviolet radiation (skin cancers) and tobacco (lung cancer). A more common source is genomic instability, which often arises when key regulatory pathways are disrupted in the cells. Some examples include impaired DNA repair mechanisms which can lead to increased replication errors, and defects in the mitosis machinery that allow for large-scale gain or loss of entire chromosomes.[32] Furthermore, it is possible for genetic variability to be further increased by some cancer therapies (e.g. treatment with temozolomide and other chemotherapy drugs).[33][34]
Mutational tumor heterogeneity refers to variations in mutation frequency in different genes and samples and can be explored by MutSig. The etiology of mutational processes can considerably vary between tumor samples from the same or different cancer types and can be manifested in different context-dependent mutational profiles. It can be explored by COSMIC mutational signatures or MutaGene.
Other heterogeneity
Tumour cells can also show heterogeneity between their expression profiles. This is often caused by underlying epigenetic changes.[31] Variation in expression signatures have been detected in different regions of tumour samples within an individual. Researchers have shown that convergent mutations affecting H3K36 methyltransferase SETD2 and histone H3K4 demethylase KDM5C arose in spatially separated tumour sections. Similarly, MTOR, a gene encoding a cell regulatory kinase, has shown to be constitutively active, thereby increasing S6 phosphorylation. This active phosphorylation may serve as a biomarker in clear-cell carcinoma.[30]
Mechanochemical heterogeneity is a hallmark of living eukaryotic cells. It has an impact on epigenetic gene regulation. The heterogeneous dynamic mechanochemical processes regulate interrelationships within the group of cellular surfaces through adhesion.[35] Tumour development and spreading is accompanied by change in heterogeneous chaotic dynamics of mechanochemical interaction process in the group cells, including cells within tumour, and is hierarchical for the host of cancer patients.[36] The biological phenomena of mechanochemical heterogeneity maybe used for differential gastric cancer diagnostics against patients with inflammation of gastric mucosa[37] and for increasing antimetastatic activity of dendritic cells based on vaccines when mechanically heterogenized microparticles of tumor cells are used for their loading.[38] There is also a possible methodical approach based on the simultaneous ultrasound imaging diagnostic techniques and therapy, regarding the mechanochemical effect on nanobubles conglomerates with drugs in the tumour.
Tumour microenvironment
Heterogeneity between tumour cells can be further increased due to heterogeneity in the tumour microenvironment. Regional differences in the tumour (e.g. availability of oxygen) impose different selective pressures on tumour cells, leading to a wider spectrum of dominant subclones in different spatial regions of the tumour. The influence of microenvironment on clonal dominance is also a likely reason for the heterogeneity between primary and metastatic tumours seen in many patients, as well as the inter-tumour heterogeneity observed between patients with the same tumour type.[39]
Implications and challenges
Treatment resistance
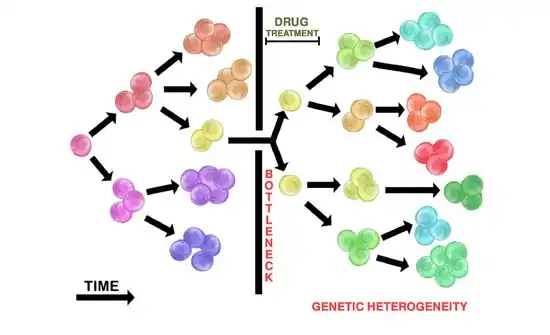
Heterogeneic tumours may exhibit different sensitivities to cytotoxic drugs among different clonal populations. This is attributed to clonal interactions that may inhibit or alter therapeutic efficacy, posing a challenge for successful therapies in heterogeneic tumours (and their heterogeneic metastases).[1]
Drug administration in heterogeneic tumours will seldom kill all tumour cells. The initial heterogeneic tumour population may bottleneck, such that few drug resistant cells (if any) will survive. This allows resistant tumour populations to replicate and grow a new tumour through the branching evolution mechanism (see above). The resulting repopulated tumour is heterogeneic and resistant to the initial drug therapy used. The repopulated tumour may also return in a more aggressive manner.
The administration of cytotoxic drugs often results in initial tumour shrinkage. This represents the destruction of initial non-resistant subclonal populations within a heterogeneic tumour, leaving only resistant clones. These resistant clones now contain a selective advantage and can replicate to repopulate the tumour. Replication will likely occur through branching evolution, contributing to tumour heterogeneity. The repopulated tumour may appear to be more aggressive. This is attributed to the drug-resistant selective advantage of the tumour cells.
Biomarker discovery
Due to the genetic differences within and between tumours, biomarkers that may predict treatment response or prognosis may not be widely applicable. However, it has been suggested that the level of heterogeneity can itself be used as a biomarker since more heterogeneous tumours may be more likely to contain treatment-resistant subclones.[31] Further research into developing biomarkers that account for heterogeneity is still in progress.
Model systems
Current model systems typically lack the heterogeneity seen in human cancers.[40] In order to accurately study tumour heterogeneity, we must develop more accurate preclinical models. One such model, the patient derived tumour xenograft, has shown excellent utility in preserving tumour heterogeneity whilst allowing detailed study of the drivers of clonal fitness.[41] However, even this model cannot capture the full complexity of cancer.
Current strategies
While the problem of identifying, characterizing, and treating tumour heterogeneity is still under active research, some effective strategies have been proposed, including both experimental and computational solutions.
Experimental
- Focused approach: analyzing a specific genetic locus or set of loci. This may occur through the detection of allelic imbalances (tumour DNA is compared to germline DNA), amplification of chromosomal regions (FISH), and/or sequencing specific genes. This method is used to trace the evolution of a specific mutation of interest, or to confirm a mutation researchers may suspect in a tumour.[1]
- Advantage: Allows for the analysis of specific genes (i.e. driver genes, tumour suppressors). The process is simple with straightforward interpretation of the results. FISH and immunofluorescence allows focus on tumour cell subtypes.[1]
- Disadvantage: Limited analysis will miss additional important mutations and patterns of clonal expansion. Allelic imbalances may be difficult to verify using microsatellite markers, therefore requiring verification by an independent technique (i.e. FISH). FISH requires large number of cells and is labour-intensive.[1]
- Genome-wide approach: analyzing the entire genome in tumour samples. This may be done through karyotyping or comparative genomic hybridization (CGH) to detect chromosomal abnormalities. Sequencing of tumour biopsies is becoming more common.[1]
- Advantage: Does not rely on prior knowledge to identify variants. karyotyping identifies regions of large chromosomal abnormalities. CGH provides unbiased coverage and allows for small-scale allelic imbalances to be detected (SNP arrays). Sequencing will identify any variants that contribute to tumour heterogeneity.[1]
- Disadvantage: Difficult to determine the functional impact of variants (i.e. neutral or pathogenic). Limited resolution. Karyotyping of cultured cells may be biased towards preferential outgrowth of select tumour cell subpopulations. Limited resolution in both methods.[1] The whole-genome approach may generate large amounts of data and be difficult to interpret.
- Multiregion sampling strategy: generally requires multiple post-surgical tumour samples from separate regions of a microdissected tumour. It is important to avoid contamination of non-malignant cells to ensure an accurate representation of gene expression and genetic composition seen within the tumour cells only. Analysis of tumour DNA within the spatially separated regions allows for the construction of a 3-dimensional evolutionary model of tumour heterogeneity.[1] Multiregional sampling is often used in combination with the genome-wide approach to establish this 3D heterogeneity expansion model.
- Longitudinal sampling: through tumour progression or treatment progression, obtaining tumour samples during multiple points in time has been utilized in some cases. This has been suggested as a reliable method for tracking clonal evolution.[34][42][43] However, this technique proves challenging in practice because it requires periodic invasive biopsy. New research into utilizing circulating cell-free tumour DNA in blood may provide a non-invasive way to identify biomarkers throughout treatment.[44] Longitudinal sampling used in combination with the genome-wide approach will allow for the identification of the accumulated tumour cell mutations through time. This may in turn identify the key driver mutations (seen in initial tumour samples).
- Adaptive therapy may be used to prevent further tumour growth by adjusting drug dose and timing of drug administration based on the tumour's response. This strategy is assumed to prevent resistant variants from dominating a tumour. However, more research is required into its applicability.[45]
Sequencing
- Bulk tumour sequencing can be utilized, where DNA is extracted from a mixture of tumour cells and analyzed all at once. The presence of heterogeneous tumour populations (subclones) introduces additional challenges such as:
- The inability to detect mutations in rare subclones. Since these mutations will occur with low frequency in the pooled sample, they may be indistinguishable from background noise. However, many variant callers are being actively developed that are specifically designed for cancer data and aim to identify rare variants present in smaller subclonal populations.[46][47][48][49] These typically utilize matched normal DNA as a means of distinguishing true somatic variation from germline variation and background sequencing error.
- The inability to determine which subclones contain each mutation. Since the data is pooled, it is not clear which mutations co-occur and which populations they originate from. New tools are being developed that attempt to resolve clonal structure using allele frequencies for the observed mutations.[50]
- Single-cell sequencing is a new technique that is valuable for assessing tumour heterogeneity because it can characterize individual tumour cells. This means that the entire mutational profile of multiple distinct cells can be determined with no ambiguity. While with current technology, it is difficult to evaluate sufficiently large numbers of single cells to obtain statistical power, single-cell tumour data has multiple advantages, including:
- The ability to construct a phylogenetic tree showing the evolution of tumour populations. Using whole-genome sequences or SNP-based pseudo-sequences from individual cells, the evolution of the subclones can be estimated. This allows for the identification of populations that have persisted over time, and can narrow down the list of mutations that potentially confer a growth advantage or treatment resistance on specific subclones.[51] Algorithms for inferring a tumor phylogeny from single-cell DNA sequencing data include SCITE,[52] OncoNEM,[53] SiFit,[54] SiCloneFit,[55] PhISCS,[56] and PhISCS-BnB.[57]
- Section sequencing can be done on multiple portions of a single solid tumour, and the variation in the mutation frequencies across the sections can be analyzed to infer the clonal structure. The advantages of this approach over single sequencing include more statistical power, and availability of more accurate information on the spatial positioning of samples. The latter can be used to infer the frequency of clones in sections and provide insight on how a tumour evolves in space. To infer the clones genotypes and phylogenetic trees that model a tumour evolution in time, several computational methods were developed[58][59][60] including Clomial,[61] cloneHD,[62] PhyloWGS,[63] PyClone,[64] Cloe,[65] phyC,[66] Canopy,[67] TargetClone, ddClone,[68] PASTRI,[69] GLClone,[70] TRaIT,[71] WSCUnmix,[72] B-SCITE.,[73] ThetA,[74] SIFA,[75] Sclust,[76] SeqClone,[77] CALDER,[78] BAMSE,[79] Meltos,[80] SubMARine,[81] and RNDCLONE.[82]
References
- Marusyk, A; Polyak, K (2010). "Tumor heterogeneity: Causes and consequences". Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 1805 (1): 105–117. doi:10.1016/j.bbcan.2009.11.002. PMC 2814927. PMID 19931353.
- Vogelstein, Bert; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. (2013). "Cancer Genome Landscapes". Science. 373 (6127): 1546–1556. Bibcode:2013Sci...339.1546V. doi:10.1126/science.1235122. PMC 3749880. PMID 23539594.
- Heppner, G.A. (1984). "Tumor Heterogeneity". Cancer Research. 44 (6): 2259–2265. PMID 6372991.
- Reiter, Johannes G; Makohon-Moore, Alvin P; Gerold, Jeffrey M; Heyde, Alexander; Attiyeh, Marc A; Kohutek, Zachary A; Tokheim, Collin J; Brown, Alexia; DeBlasio, Rayne; Niyazov, Juliana; Zucker, Amanda; Karchin, Rachel; Kinzler, Kenneth W; Iacobuzio-Donahue, Christine A; Vogelstein, Bert; Nowak, Martin A (2018). "Minimal functional driver gene heterogeneity among untreated metastases". Science. 361 (6406): 1033–1037. doi:10.1126/science.aat7171. PMC 6329287. PMID 30190408.
- Campbell, P. J.; Pleasance, E. D.; Stephens, P. J.; Dicks, E; Rance, R; Goodhead, I; Follows, G. A.; Green, A. R.; Futreal, P. A.; Stratton, M. R. (2008). "Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing". Proceedings of the National Academy of Sciences. 105 (35): 13081–13086. Bibcode:2008PNAS..10513081C. doi:10.1073/pnas.0801523105. PMC 2529122. PMID 18723673.
- Shipitsin, M; Campbell, L. L.; Argani, P; Weremowicz, S; Bloushtain-Qimron, N; Yao, J; Nikolskaya, T; Serebryiskaya, T; Beroukhim, R; Hu, M; Halushka, M. K.; Sukumar, S; Parker, L. M.; Anderson, K. S.; Harris, L. N.; Garber, J. E.; Richardson, A. L.; Schnitt, S. J.; Nikolsky, Y; Gelman, R. S.; Polyak, K (2007). "Molecular definition of breast tumor heterogeneity". Cancer Cell. 11 (3): 259–273. doi:10.1016/j.ccr.2007.01.013. PMID 17349583.
- MacIntosh, C. A.; Stower, M; Reid, N; Maitland, N. J. (1998). "Precise microdissection of human prostate cancers reveals genotypic heterogeneity". Cancer Research. 58 (1): 23–28. PMID 9426051.
- Alvarado, C; Beitel, L. K.; Sircar, K; Aprikian, A; Trifiro, M; Gottlieb, B (2005). "Somatic mosaicism and cancer: A micro-genetic examination into the role of the androgen receptor gene in prostate cancer". Cancer Research. 65 (18): 8514–8518. doi:10.1158/0008-5472.CAN-05-0399. PMID 16166332.
- Konishi, N; Hiasa, Y; Matsuda, H; Tao, M; Tsuzuki, T; Hayashi, I; Kitahori, Y; Shiraishi, T; Yatani, R; Shimazaki, J (1995). "Intratumor cellular heterogeneity and alterations in ras oncogene and p53 tumor suppressor gene in human prostate carcinoma". The American Journal of Pathology. 147 (4): 1112–1122. PMC 1871010. PMID 7573356.
- González-García, I; Solé, R. V.; Costa, J (2002). "Metapopulation dynamics and spatial heterogeneity in cancer". Proceedings of the National Academy of Sciences. 99 (20): 13085–13089. Bibcode:2002PNAS...9913085G. doi:10.1073/pnas.202139299. PMC 130590. PMID 12351679.
- Samowitz, W. S.; Slattery, M. L. (1999). "Regional reproducibility of microsatellite instability in sporadic colorectal cancer". Genes, Chromosomes and Cancer. 26 (2): 106–114. doi:10.1002/(SICI)1098-2264(199910)26:2<106::AID-GCC2>3.0.CO;2-F. PMID 10469448.
- Giaretti, W; Monaco, R; Pujic, N; Rapallo, A; Nigro, S; Geido, E (1996). "Intratumor heterogeneity of K-ras2 mutations in colorectal adenocarcinomas: Association with degree of DNA aneuploidy". The American Journal of Pathology. 149 (1): 237–245. PMC 1865212. PMID 8686748.
- Heppner, G. H. (1984). "Tumor heterogeneity". Cancer Research. 44 (6): 2259–2265. PMID 6372991.
- Maley, C. C.; Galipeau, P. C.; Finley, J. C.; Wongsurawat, V. J.; Li, X; Sanchez, C. A.; Paulson, T. G.; Blount, P. L.; Risques, R. A.; Rabinovitch, P. S.; Reid, B. J. (2006). "Genetic clonal diversity predicts progression to esophageal adenocarcinoma". Nature Genetics. 38 (4): 468–473. doi:10.1038/ng1768. PMID 16565718.
- Califano, J; Van Der Riet, P; Westra, W; Nawroz, H; Clayman, G; Piantadosi, S; Corio, R; Lee, D; Greenberg, B; Koch, W; Sidransky, D (1996). "Genetic progression model for head and neck cancer: Implications for field cancerization". Cancer Research. 56 (11): 2488–2492. PMID 8653682.
- Sauter, G; Moch, H; Gasser, T. C.; Mihatsch, M. J.; Waldman, F. M. (1995). "Heterogeneity of chromosome 17 and erbB-2 gene copy number in primary and metastatic bladder cancer". Cytometry. 21 (1): 40–46. doi:10.1002/cyto.990210109. PMID 8529469.
- Fujii, H; Yoshida, M; Gong, Z. X.; Matsumoto, T; Hamano, Y; Fukunaga, M; Hruban, R. H.; Gabrielson, E; Shirai, T (2000). "Frequent genetic heterogeneity in the clonal evolution of gynecological carcinosarcoma and its influence on phenotypic diversity". Cancer Research. 60 (1): 114–120. PMID 10646862.
- Horvai, A. E.; Devries, S; Roy, R; O'Donnell, R. J.; Waldman, F (2009). "Similarity in genetic alterations between paired well-differentiated and dedifferentiated components of dedifferentiated liposarcoma". Modern Pathology. 22 (11): 1477–1488. doi:10.1038/modpathol.2009.119. PMID 19734852.
- Pantou, D; Rizou, H; Tsarouha, H; Pouli, A; Papanastasiou, K; Stamatellou, M; Trangas, T; Pandis, N; Bardi, G (2005). "Cytogenetic manifestations of multiple myeloma heterogeneity". Genes, Chromosomes and Cancer. 42 (1): 44–57. doi:10.1002/gcc.20114. PMID 15495197.
- Shackleton, M; Quintana, E; Fearon, E. R.; Morrison, S. J. (2009). "Heterogeneity in cancer: Cancer stem cells versus clonal evolution". Cell. 138 (5): 822–829. doi:10.1016/j.cell.2009.08.017. PMID 19737509.
- Lapidot, T; Sirard, C; Vormoor, J; Murdoch, B; Hoang, T; Caceres-Cortes, J; Minden, M; Paterson, B; Caligiuri, M. A.; Dick, J. E. (1994). "A cell initiating human acute myeloid leukaemia after transplantation into SCID mice". Nature. 367 (6464): 645–648. Bibcode:1994Natur.367..645L. doi:10.1038/367645a0. PMID 7509044.
- Wang, J. C.; Lapidot, T; Cashman, J. D.; Doedens, M; Addy, L; Sutherland, D. R.; Nayar, R; Laraya, P; Minden, M; Keating, A; Eaves, A. C.; Eaves, C. J.; Dick, J. E. (1998). "High level engraftment of NOD/SCID mice by primitive normal and leukemic hematopoietic cells from patients with chronic myeloid leukemia in chronic phase". Blood. 91 (7): 2406–2414. doi:10.1182/blood.V91.7.2406. PMID 9516140.
- Singh, S. K.; Hawkins, C; Clarke, I. D.; Squire, J. A.; Bayani, J; Hide, T; Henkelman, R. M.; Cusimano, M. D.; Dirks, P. B. (2004). "Identification of human brain tumour initiating cells". Nature. 432 (7015): 396–401. Bibcode:2004Natur.432..396S. doi:10.1038/nature03128. PMID 15549107.
- Al-Hajj, M; Wicha, M. S.; Benito-Hernandez, A; Morrison, S. J.; Clarke, M. F. (2003). "Prospective identification of tumorigenic breast cancer cells". Proceedings of the National Academy of Sciences. 100 (7): 3983–3988. Bibcode:2003PNAS..100.3983A. doi:10.1073/pnas.0530291100. PMC 153034. PMID 12629218.
- Maitland, N. J.; Collins, A. T. (2008). "Prostate cancer stem cells: A new target for therapy". Journal of Clinical Oncology. 26 (17): 2862–2870. doi:10.1200/JCO.2007.15.1472. PMID 18539965.
- Meacham, C. E.; Morrison, S. J. (2013). "Tumour heterogeneity and cancer cell plasticity". Nature. 501 (7467): 328–337. Bibcode:2013Natur.501..328M. doi:10.1038/nature12624. PMC 4521623. PMID 24048065.
- Nowell, P. C. (1976). "The clonal evolution of tumor cell populations". Science. 194 (4260): 23–28. Bibcode:1976Sci...194...23N. doi:10.1126/science.959840. PMID 959840.
- Swanton, C (2012). "Intratumor heterogeneity: Evolution through space and time". Cancer Research. 72 (19): 4875–4882. doi:10.1158/0008-5472.CAN-12-2217. PMC 3712191. PMID 23002210.
- Merlo, L. M. F.; Pepper, J. W.; Reid, B. J.; Maley, C. C. (2006). "Cancer as an evolutionary and ecological process". Nature Reviews Cancer. 6 (12): 924–935. doi:10.1038/nrc2013. PMID 17109012.
- Gerlinger, M; Rowan, A. J.; Horswell, S; Larkin, J; Endesfelder, D; Gronroos, E; Martinez, P; Matthews, N; Stewart, A; Tarpey, P; Varela, I; Phillimore, B; Begum, S; McDonald, N. Q.; Butler, A; Jones, D; Raine, K; Latimer, C; Santos, C. R.; Nohadani, M; Eklund, A. C.; Spencer-Dene, B; Clark, G; Pickering, L; Stamp, G; Gore, M; Szallasi, Z; Downward, J; Futreal, P. A.; Swanton, C (2012). "Intratumor heterogeneity and branched evolution revealed by multiregion sequencing". New England Journal of Medicine. 366 (10): 883–892. doi:10.1056/NEJMoa1113205. PMC 4878653. PMID 22397650.
- Marusyk, A; Almendro, V; Polyak, K (2012). "Intra-tumour heterogeneity: A looking glass for cancer?". Nature Reviews Cancer. 12 (5): 323–334. doi:10.1038/nrc3261. PMID 22513401.
- Burrell, R. A.; McGranahan, N; Bartek, J; Swanton, C (2013). "The causes and consequences of genetic heterogeneity in cancer evolution". Nature. 501 (7467): 338–345. Bibcode:2013Natur.501..338B. doi:10.1038/nature12625. PMID 24048066.
- Johnson, B. E.; Mazor, T; Hong, C; Barnes, M; Aihara, K; McLean, C. Y.; Fouse, S. D.; Yamamoto, S; Ueda, H; Tatsuno, K; Asthana, S; Jalbert, L. E.; Nelson, S. J.; Bollen, A. W.; Gustafson, W. C.; Charron, E; Weiss, W. A.; Smirnov, I. V.; Song, J. S.; Olshen, A. B.; Cha, S; Zhao, Y; Moore, R. A.; Mungall, A. J.; Jones, S. J.; Hirst, M; Marra, M. A.; Saito, N; Aburatani, H; Mukasa, A (2014). "Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma". Science. 343 (6167): 189–193. Bibcode:2014Sci...343..189J. doi:10.1126/science.1239947. PMC 3998672. PMID 24336570.
- Ding, L; Ley, T. J.; Larson, D. E.; Miller, C. A.; Koboldt, D. C.; Welch, J. S.; Ritchey, J. K.; Young, M. A.; Lamprecht, T; McLellan, M. D.; McMichael, J. F.; Wallis, J. W.; Lu, C; Shen, D; Harris, C. C.; Dooling, D. J.; Fulton, R. S.; Fulton, L. L.; Chen, K; Schmidt, H; Kalicki-Veizer, J; Magrini, V. J.; Cook, L; McGrath, S. D.; Vickery, T. L.; Wendl, M. C.; Heath, S; Watson, M. A.; Link, D. C.; Tomasson, M. H. (2012). "Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing". Nature. 481 (7382): 506–510. Bibcode:2012Natur.481..506D. doi:10.1038/nature10738. PMC 3267864. PMID 22237025.
- G.M.Edelman (1989). "Topobiology". J. Scientific American. 260 (5): 76–88. doi:10.1038/scientificamerican0589-76. PMID 2717916.
- V.E. Orel; N.N Dzyatkovskaya; M.I. Danko; A.V. Romanov; Y.I. Mel'nik; Y.A. Grinevich; S.V. Martynenko (2004). "Spatial and mechanoemission chaos of mechanically deformed tumor cells". J. Journal of Mechanics in Medicine and Biology. 4 (1): 31–45. doi:10.1142/s0219519404000886.
- V.E. Orel; A.V. Romanov; N.N. Dzyatkovskaya; Yu.I. Mel’nik (2002). "The device and algorithm for estimation of the mechanoemisson chaos in blood of patients with gastric cancer". J. Medical Engineering & Physics. 24 (5): 365–371. doi:10.1016/s1350-4533(02)00022-x. PMID 12052364.
- N. Khranovskaya; V. Orel; Y. Grinevich; O. Alekseenko; A. Romanov; O. Skachkova; N.Dzyatkovskaya; A. Burlaka; S.Lukin (2012). "Mechanical heterogenization of Lewis lung carcinoma cells can improve antimetastatic effect of dendritic cells". J. Journal of Mechanics in Medicine and Biology. 3 (12): 22. doi:10.1142/S0219519411004757.
- Junttila, M. R.; De Sauvage, F. J. (2013). "Influence of tumour micro-environment heterogeneity on therapeutic response". Nature. 501 (7467): 346–354. Bibcode:2013Natur.501..346J. doi:10.1038/nature12626. PMID 24048067.
- Auman, James Todd; McLeod, Howard L. (2010-01-01). "Colorectal Cancer Cell Lines Lack the Molecular Heterogeneity of Clinical Colorectal Tumors". Clinical Colorectal Cancer. 9 (1): 40–47. doi:10.3816/ccc.2010.n.005. PMID 20100687.
- Cassidy, John W.; Caldas, Carlos; Bruna, Alejandra (2015-08-01). "Maintaining Tumor Heterogeneity in Patient-Derived Tumor Xenografts". Cancer Research. 75 (15): 2963–2968. doi:10.1158/0008-5472.CAN-15-0727. ISSN 0008-5472. PMC 4539570. PMID 26180079.
- Bai H, Harmancı AS, Erson-Omay AZ, Li J, Coșkun S, Simon M, et al. (Nov 2015). "Integrated genomic characterization of IDH1-mutant glioma malignant progression". Nature Genetics. 48 (1): 59–66. doi:10.1038/ng.3457. PMC 4829945. PMID 26618343.
- Bedard, P. L.; Hansen, A. R.; Ratain, M. J.; Siu, L. L. (2013). "Tumour heterogeneity in the clinic". Nature. 501 (7467): 355–364. Bibcode:2013Natur.501..355B. doi:10.1038/nature12627. PMC 5224525. PMID 24048068.
- Dawson, S. J.; Tsui, D. W.; Murtaza, M; Biggs, H; Rueda, O. M.; Chin, S. F.; Dunning, M. J.; Gale, D; Forshew, T; Mahler-Araujo, B; Rajan, S; Humphray, S; Becq, J; Halsall, D; Wallis, M; Bentley, D; Caldas, C; Rosenfeld, N (2013). "Analysis of circulating tumor DNA to monitor metastatic breast cancer". New England Journal of Medicine. 368 (13): 1199–1209. doi:10.1056/NEJMoa1213261. PMID 23484797.
- Gatenby, R. A.; Silva, A. S.; Gillies, R. J.; Frieden, B. R. (2009). "Adaptive therapy". Cancer Research. 69 (11): 4894–4903. doi:10.1158/0008-5472.CAN-08-3658. PMC 3728826. PMID 19487300.
- Cibulskis, K; Lawrence, M. S.; Carter, S. L.; Sivachenko, A; Jaffe, D; Sougnez, C; Gabriel, S; Meyerson, M; Lander, E. S.; Getz, G (2013). "Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples". Nature Biotechnology. 31 (3): 213–219. doi:10.1038/nbt.2514. PMC 3833702. PMID 23396013.
- Koboldt, D. C.; Zhang, Q; Larson, D. E.; Shen, D; McLellan, M. D.; Lin, L; Miller, C. A.; Mardis, E. R.; Ding, L; Wilson, R. K. (2012). "Var Scan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing". Genome Research. 22 (3): 568–576. doi:10.1101/gr.129684.111. PMC 3290792. PMID 22300766.
- Saunders, C. T.; Wong, W. S.; Swamy, S; Becq, J; Murray, L. J.; Cheetham, R. K. (2012). "Strelka: Accurate somatic small-variant calling from sequenced tumor-normal sample pairs". Bioinformatics. 28 (14): 1811–1817. doi:10.1093/bioinformatics/bts271. PMID 22581179.
- Carter, S. L.; Cibulskis, K; Helman, E; McKenna, A; Shen, H; Zack, T; Laird, P. W.; Onofrio, R. C.; Winckler, W; Weir, B. A.; Beroukhim, R; Pellman, D; Levine, D. A.; Lander, E. S.; Meyerson, M; Getz, G (2012). "Absolute quantification of somatic DNA alterations in human cancer" (PDF). Nature Biotechnology. 30 (5): 413–421. doi:10.1038/nbt.2203. PMC 4383288. PMID 22544022.
- Shah, S. P.; Roth, A; Goya, R; Oloumi, A; Ha, G; Zhao, Y; Turashvili, G; Ding, J; Tse, K; Haffari, G; Bashashati, A; Prentice, L. M.; Khattra, J; Burleigh, A; Yap, D; Bernard, V; McPherson, A; Shumansky, K; Crisan, A; Giuliany, R; Heravi-Moussavi, A; Rosner, J; Lai, D; Birol, I; Varhol, R; Tam, A; Dhalla, N; Zeng, T; Ma, K; Chan, S. K. (2012). "The clonal and mutational evolution spectrum of primary triple-negative breast cancers". Nature. 486 (7403): 395–399. Bibcode:2012Natur.486..395S. doi:10.1038/nature10933. PMC 3863681. PMID 22495314.
- Navin, N; Kendall, J; Troge, J; Andrews, P; Rodgers, L; McIndoo, J; Cook, K; Stepansky, A; Levy, D; Esposito, D; Muthuswamy, L; Krasnitz, A; McCombie, W. R.; Hicks, J; Wigler, M (2011). "Tumour evolution inferred by single-cell sequencing". Nature. 472 (7341): 90–94. Bibcode:2011Natur.472...90N. doi:10.1038/nature09807. PMC 4504184. PMID 21399628.
- Jahn, Katharina (2016). "Tree inference for single-cell data". Genome Biology. 17: 86. doi:10.1186/s13059-016-0936-x. PMC 4858868. PMID 27149953.
- Ross, Edith (2016). "OncoNEM: inferring tumor evolution from single-cell sequencing data". Genome Biology. 17: 69. doi:10.1186/s13059-016-0929-9. PMC 4832472. PMID 27083415.
- Zafar, Hamim (2017). "SiFit: inferring tumor trees from single-cell sequencing data under finite-sites models". Genome Biology. 18 (1): 178. doi:10.1186/s13059-017-1311-2. PMC 5606061. PMID 28927434.
- Zafar, Hamim (2019). "SiCloneFit: Bayesian inference of population structure, genotype, and phylogeny of tumor clones from single-cell genome sequencing data". Genome Research. 29 (11): 1847–1859. doi:10.1101/gr.243121.118. PMC 6836738. PMID 31628257.
- Malikic, Salem; Rashidi Mehrabadi, Farid (2019). "PhISCS: a combinatorial approach for subperfect tumor phylogeny reconstruction via integrative use of single-cell and bulk sequencing data". Genome Research. 29 (11): 1860–1877. doi:10.1101/gr.234435.118. PMID 31628256.
- Sadeqi Azer, Erfan; Rashidi Mehrabadi, Farid (2020). "PhISCS-BnB: a fast branch and bound algorithm for the perfect tumor phylogeny reconstruction problem". Bioinformatics. 36 (Supplement_1): i169–i176. doi:10.1093/bioinformatics/btaa464. PMID 32657358.
- Kuipers, Jack (2017). "Advances in understanding tumour evolution through single-cell sequencing". Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 1867 (2): 127–138. doi:10.1016/j.bbcan.2017.02.001. PMC 5813714. PMID 28193548.
- Schwartz, Russell (13 Feb 2017). "The evolution of tumour phylogenetics: principles and practice". Nature Reviews Genetics. 18 (4): 213–229. doi:10.1038/nrg.2016.170. PMC 5886015. PMID 28190876.
- Farahani, Hossein; de Souza, Camila P. E.; Billings, Raewyn; Yap, Damian; Shumansky, Karey; Wan, Adrian; Lai, Daniel; Mes-Masson, Anne-Marie; Aparicio, Samuel; P. Shah, Sohrab (18 October 2017). "Engineered in-vitro cell line mixtures and robust evaluation of computational methods for clonal decomposition and longitudinal dynamics in cancer". Scientific Reports. 7 (1): 13467. Bibcode:2017NatSR...713467F. doi:10.1038/s41598-017-13338-8. PMC 5647443. PMID 29044127.
- Zare, Habil (2014). "Inferring clonal composition from multiple sections of a breast cancer". PLOS Computational Biology. 10 (7): e1003703. Bibcode:2014PLSCB..10E3703Z. doi:10.1371/journal.pcbi.1003703. PMC 4091710. PMID 25010360.
- Fischer, Andrej (2014). "High-definition reconstruction of clonal composition in cancer". Cell Reports. 7 (5): 1740–1752. doi:10.1016/j.celrep.2014.04.055. PMC 4062932. PMID 24882004.
- Deshwar, Amit (2015). "Monitoring chronic lymphocytic leukemia progression by whole genome sequencing reveals heterogeneous clonal evolution patterns". Genome Biology. 16: 35. doi:10.1186/s13059-015-0602-8. PMC 4359439. PMID 25786235.
- Roth, Andrew (2014). "PyClone: statistical inference of clonal population structure in cancer". Nature Methods. 11 (4): 396–398. doi:10.1038/nmeth.2883. PMC 4864026. PMID 24633410.
- Marass, Francesco (2015). "A phylogenetic latent feature model for clonal deconvolution". The Annals of Applied Statistics. 10 (4): 2377–2404. arXiv:1604.01715. doi:10.1214/16-AOAS986.
- Matsui, Yusuke (2016). "phyC: Clustering cancer evolutionary trees". PLOS Computational Biology. 13 (5): e1005509. Bibcode:2017PLSCB..13E5509M. bioRxiv 10.1101/069302. doi:10.1371/journal.pcbi.1005509. PMC 5432190. PMID 28459850.
- Jiang, Yuchao; Qiu, Yu; Minn, Andy J.; Zhang, Nancy R. (29 August 2016). "Assessing intratumor heterogeneity and tracking longitudinal and spatial clonal evolutionary history by next-generation sequencing". Proceedings of the National Academy of Sciences. 113 (37): E5528–37. doi:10.1073/pnas.1522203113. PMC 5027458. PMID 27573852.
- Salehi, Sohrab (2017). "ddClone: joint statistical inference of clonal populations from single cell and bulk tumour sequencing data". Genome Biology. 18 (1): 44. doi:10.1186/s13059-017-1169-3. PMC 5333399. PMID 28249593.
- Satas, Gryte (2017). "Tumor phylogeny inference using tree-constrained importance sampling". Bioinformatics. 33 (14): i152–i160. doi:10.1093/bioinformatics/btx270. PMC 5870673. PMID 28882002.
- Geng, Yu (2017). "Identifying Heterogeneity Patterns of Allelic Imbalance on Germline Variants to Infer Clonal Architecture". International Conference on Intelligent Computing. Lecture Notes in Computer Science. 10362: 286–297. doi:10.1007/978-3-319-63312-1_26. ISBN 978-3-319-63311-4.
- Ramazzotti, Daniele; Graudenzi, Alex; Sano, Luca De; Antoniotti, Marco; Caravagna, Giulio (4 September 2017). "Learning mutational graphs of individual tumor evolution from multi-sample sequencing data". bioRxiv 10.1101/132183.
- Roman, Theodore; Xie, Lu; Schwartz, Russell; Raphael, Benjamin J. (23 October 2017). "Automated deconvolution of structured mixtures from heterogeneous tumor genomic data". PLOS Computational Biology. 13 (10): e1005815. arXiv:1604.02487. Bibcode:2017PLSCB..13E5815R. doi:10.1371/journal.pcbi.1005815. PMC 5695636. PMID 29059177.
- Malikic, Salem (2017). "Integrative inference of subclonal tumour evolution from single-cell and bulk sequencing data". bioRxiv 10.1101/234914.
- Oesper, Layla; Mahmoody, Ahmad; Raphael, Benjamin J. (29 July 2013). "THetA: inferring intra-tumor heterogeneity from high-throughput DNA sequencing data". Genome Biology. 14 (7): R80. doi:10.1186/gb-2013-14-7-r80. ISSN 1474-760X. PMC 4054893. PMID 23895164.
- Zeng, By (2018). "Phylogeny-based tumor subclone identification using a Bayesian feature allocation model". arXiv:1803.06393 [stat.AP].
- Cun, Yupeng; Yang, Tsun-Po; Achter, Viktor; Lang, Ulrich; Peifer, Martin (24 May 2018). "Copy-number analysis and inference of subclonal populations in cancer genomes using Sclust". Nature Protocols. 13 (6): 1488–1501. doi:10.1038/nprot.2018.033. ISSN 1754-2189. PMID 29844525.
- Wang, Xiaodong; Ogundijo, Oyetunji E. (2019-12-01). "SeqClone: sequential Monte Carlo based inference of tumor subclones". BMC Bioinformatics. 20 (1): 6. doi:10.1186/s12859-018-2562-y. ISSN 1471-2105. PMC 6320595. PMID 30611189.
- Raphael, Benjamin J.; Satas, Gryte; Myers, Matthew A. (22 January 2019). "Inferring tumor evolution from longitudinal samples". bioRxiv: 526814. doi:10.1101/526814.
- Toosi, Hosein; Moeini, Ali; Hajirasouliha, Iman (6 June 2019). "BAMSE: Bayesian model selection for tumor phylogeny inference among multiple samples". BMC Bioinformatics. 20 (11): 282. doi:10.1186/s12859-019-2824-3. ISSN 1471-2105. PMC 6551234. PMID 31167637.
- Ricketts, Camir; Seidman, Daniel; Popic, Victoria; Hormozdiari, Fereydoun; Batzoglou, Serafim; Hajirasouliha, Iman (4 October 2019). "Meltos: Multi-Sample Tumor Phylogeny Reconstruction for Structural Variants". Bioinformatics. 36 (4): 1082–1090. doi:10.1093/bioinformatics/btz737. PMID 31584621.
- Sundermann, Linda. "Reconstructing tumor evolutionary histories and clone trees in polynomial-time with SubMARine" (PDF). bioRxiv. Retrieved 22 June 2020.
- Zhou, Tianjian. "RNDCLONE: TUMOR SUBCLONE RECONSTRUCTION BASED ON INTEGRATING DNA AND RNA SEQUENCE DATA" (PDF). Retrieved 23 August 2020.