Rubber elasticity
Rubber elasticity refers to a property of crosslinked rubber: it can be stretched by up to a factor of 10 from its original length and, when released, returns very nearly to its original length. This can be repeated many times with no apparent degradation to the rubber. Rubber is a member of a larger class of materials called elastomers and it is difficult to overestimate their economic and technological importance. Elastomers have played a key role in the development of new technologies in the 20th century and make a substantial contribution to the global economy. Rubber elasticity is produced by several complex molecular processes and its explanation requires a knowledge of advanced mathematics, chemistry and statistical physics, particularly the concept of entropy. Entropy may be thought of as a measure of the thermal energy that is stored in a molecule. Common rubbers, such as polybutadiene and polyisoprene (also called natural rubber), are produced by a process called polymerization. Very long molecules (polymers) are built up sequentially by adding short molecular backbone units through chemical reactions. A rubber polymer follows a random, zigzag path in three dimensions, intermingling with many other rubber molecules. An elastomer is created by the addition of a few percent of a cross linking molecule such as sulfur. When heated, the crosslinking molecule causes a reaction that chemically joins (bonds) two of the rubber molecules together at some point (a crosslink). Because the rubber molecules are so long, each one participates in many crosslinks with many other rubber molecules forming a continuous molecular network. As a rubber band is stretched, some of the network chains are forced to become straight and this causes a decrease in their entropy. It is this decrease in entropy that gives rise to the elastic force in the network chains.
History
Following its introduction to Europe from the New World in the late 15th century, natural rubber (polyisoprene) was regarded mostly as a fascinating curiosity. Its most useful application was its ability to erase pencil marks on paper by rubbing, hence its name. One of its most peculiar properties is a slight (but detectable) increase in temperature that occurs when a sample of rubber is stretched. If it is allowed to quickly retract, an equal amount of cooling is observed. This phenomenon caught the attention of the English physicist John Gough. In 1805 he published some qualitative observations on this characteristic as well as how the required stretching force increased with temperature.[1]
By the mid nineteenth century, the theory of thermodynamics was being developed and within this framework, the English mathematician and physicist Lord Kelvin[2] showed that the change in mechanical energy required to stretch a rubber sample should be proportional to the increase in temperature. Later, this would be associated with a change in entropy. The connection to thermodynamics was firmly established in 1859 when the English physicist James Joule published the first careful measurements of the temperature increase that occurred as a rubber sample was stretched.[3] This work confirmed the theoretical predictions of Lord Kelvin.
It was not until 1838 that the American inventor Charles Goodyear found that natural rubber's properties could be immensely improved by adding a few percent sulphur. The short sulfur chains produced chemical cross-links between adjacent polyisoprene molecules. Before it is cross-linked, the liquid natural rubber consists of very long linear chains, containing thousands of isoprene backbone units, connected head-to-tail. Every chain follows a random path through the liquid and is in contact with thousands of other nearby chains. When heated to about 150C, cross-linker molecules (such as sulfur or dicumyl peroxide) can decompose and the subsequent chemical reactions produce a chemical bond between adjacent chains. The result is a three dimensional molecular network. All of the original polyisoprene chains are connected together at multiple points by these chemical bonds (network nodes) to form a single giant molecule. A rubber band is a single molecule, as is a latex glove! The sections between two cross-links on the same chain are called network chains and can contain up to several hundred isoprene units. In natural rubber, each cross-link produces a network node with four chains emanating from it. The network is the sine qua non of elastomers.
Because of the enormous economic and technological importance of rubber, predicting how a molecular network responds to mechanical strains has been of enduring interest to scientists and engineers. To understand the elastic properties of rubber, theoretically, it is necessary to know both the physical mechanisms that occur at the molecular level and how the random-walk nature of the polymer chain defines the network. The physical mechanisms that occur within short sections of the polymer chains produce the elastic forces and the network morphology determines how these forces combine to produce the macroscopic stress that we observe when a rubber sample is deformed, e.g. subjected to tensile strain.
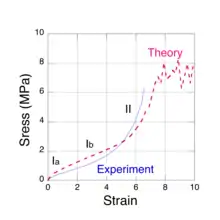
Molecular-level models
There are actually several physical mechanisms that produce the elastic forces within the network chains as a rubber sample is stretched. Two of these arise from entropy changes and one is associated with the distortion of the molecular bond angles along the chain backbone. These three mechanisms are immediately apparent when a moderately thick rubber sample is stretched manually. Initially, the rubber feels quite stiff, i.e. the force must be increased at a high rate with respect to the strain. At intermediate strains, the required increase in force is much lower to cause the same amount of stretch. Finally, as the sample approaches the breaking point, its stiffness increases markedly. What the observer is noticing are the changes in the modulus of elasticity that are due to the different molecular mechanisms. These regions can be seen in Fig. 1, a typical stress vs. strain measurement for natural rubber. The three mechanisms (labelled Ia, Ib and II) predominantly correspond to the regions shown on the plot. The concept of entropy comes to us from the area mathematical physics called statistical mechanics which is concerned with the study of large thermal systems, e.g. rubber networks at room temperature. Although the detailed behavior of the constituent chains are random and far too complex to study individually, we can obtain very useful information about their 'average' behavior from a statistical mechanics analysis of a large sample. There are no other examples of how entropy changes can produce a force in our everyday experience. One may regard the entropic forces in polymer chains as arising from the thermal collisions that their constituent atoms experience with the surrounding material. It is this constant jostling that produces a resisting (elastic) force in the chains as they are forced to become straight. While stretching a rubber sample is the most common example of elasticity, it also occurs when rubber is compressed. Compression may be thought of as a two dimensional expansion as when a balloon is inflated. The molecular mechanisms that produce the elastic force are the same for all types of strain.
When these elastic force models are combined with the complex morphology of the network, it is not possible to obtain simple analytic formulae to predict the macroscopic stress. It is only via numerical simulations on computers that it is possible to capture the complex interaction between the molecular forces and the network morphology to predict the stress and ultimate failure of a rubber sample as it is strained.
The Molecular Kink Paradigm for rubber elasticity[4]
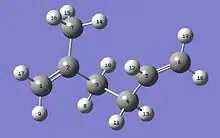
The Molecular Kink Paradigm proceeds from the intuitive notion that molecular chains that make up a natural rubber (polyisoprene) network are constrained by surrounding chains to remain within a ‘tube’. Elastic forces produced in a chain, as a result of some applied strain, are propagated along the chain contour within this tube. Fig. 2 shows a representation of a four-carbon isoprene backbone unit with an extra carbon atom at each end to indicate its connections to adjacent units on a chain. It has three single C-C bonds and one double bond. It is principally by rotating about the C-C single bonds that a polyisoprene chain randomly explores its possible conformations. Sections of chain containing between two and three isoprene units have sufficient flexibility that they may be considered statistically de-correlated from one another. That is, there is no directional correlation along the chain for distances greater than this distance, referred to as a Kuhn length. These non-straight regions evoke the concept of ‘kinks’ and are in fact a manifestation of the random-walk nature of the chain. Since a kink is composed of several isoprene units, each having three carbon-carbon single bonds, there are many possible conformations available to a kink, each with a distinct energy and end-to-end distance. Over time scales of seconds to minutes, only these relatively short sections of the chain, i.e. kinks, have sufficient volume to move freely amongst their possible rotational conformations. The thermal interactions tend to keep the kinks in a state of constant flux, as they make transitions between all of their possible rotational conformations. Because the kinks are in thermal equilibrium, the probability that a kink resides in any rotational conformation is given by a Boltzmann distribution and we may associate an entropy with its end-to-end distance. The probability distribution for the end-to-end distance of a Kuhn length is approximately Gaussian and is determined by the Boltzmann probability factors for each state (rotational conformation). As a rubber network is stretched, some kinks are forced into a restricted number of more extended conformations having a greater end-to-end distance and it is the resulting decrease in entropy that produces an elastic force along the chain.
There are three distinct molecular mechanisms that produce these forces, two of which arise from changes in entropy that we shall refer to as low chain extension regime, Ia[5] and moderate chain extension regime, Ib.[6] The third mechanism occurs at high chain extension, as it is extended beyond its initial equilibrium contour length by the distortion of the chemical bonds along its backbone. In this case, the restoring force is spring-like and we shall refer to it as regime II.[7] The three force mechanisms are found to roughly correspond to the three regions observed in tensile stress vs. strain experiments, shown in Fig. 1.
The initial morphology of the network, immediately after chemical cross-linking, is governed by two random processes:[8][9] (1) The probability for a cross-link to occur at any isoprene unit and, (2) the random walk nature of the chain conformation. The end-to-end distance probability distribution for a fixed chain length, i.e. fixed number of isoprene units, is described by a random walk. It is the joint probability distribution of the network chain lengths and the end-to-end distances between their cross-link nodes that characterizes the network morphology. Because both the molecular physics mechanisms that produce the elastic forces and the complex morphology of the network must be treated simultaneously, simple analytic elasticity models are not possible; an explicit 3-dimensional numerical model[10][11][12] is required to simulate the effects of strain on a representative volume element of a network.
Low chain extension regime, Ia
The Molecular Kink Paradigm envisions a representative network chain as a series of vectors that follow the chain contour within its tube. Each vector represents the equilibrium end-to-end distance of a kink. The actual 3-dimensional path of the chain is not pertinent, since all elastic forces are assumed to operate along the chain contour. In addition to the chain's contour length, the only other important parameter is its tortuosity, the ratio of its contour length to its end-to-end distance. As the chain is extended, in response to an applied strain, the induced elastic force is assumed to propagate uniformly along its contour. Consider a network chain whose end points (network nodes) are more or less aligned with the tensile strain axis. As the initial strain is applied to the rubber sample, the network nodes at the ends of the chain begin to move apart and all of the kink vectors along the contour are stretched simultaneously. Physically, the applied strain forces the kinks to stretch beyond their thermal equilibrium end-to-end distances, causing a decrease in their entropy. The increase in free energy associated with this change in entropy, gives rise to a (linear) elastic force that opposes the strain. The force constant for the low strain regime can be estimated by sampling molecular dynamics (MD) trajectories of a kink, i.e. short chains, composed of 2-3 isoprene units, at relevant temperatures, e.g. 300K.[5] By taking many samples of the coordinates over the course of the simulations, the probability distributions of end-to-end distance for a kink can be obtained. Since these distributions (which turn out to be approximately Gaussian) are directly related to the number of states, we may associate them with the entropy of the kink at any end-to-end distance. By numerically differentiating the probability distribution, the change in entropy, and hence free energy, with respect to the kink end-to-end distance can be found. The force model for this regime is found to be linear and proportional to the temperature divided by the chain tortuosity.
Moderate chain extension regime, Ib
At some point in the low extension regime, i.e. as all of the kinks along the chain are being extended simultaneously, it becomes energetically more favorable to have one kink transition to an extended conformation in order to stretch the chain further. The applied strain can force a single isoprene unit within a kink into an extended conformation, slightly increasing the end-to-end distance of the chain, and the energy required to do this is less than that needed to continue extending all of the kinks simultaneously. Numerous experiments[13] strongly suggest that stretching a rubber network is accompanied by a decrease in entropy. As shown in Fig. 2, an isoprene unit has three single C-C bonds and there are two or three preferred rotational angles (orientations) about these bonds that have energy minima. Of the18 allowed[6] rotational conformations, only 6 have extended end-to-end distances and forcing the isoprene units in a chain to reside in some subset of the extended states must reduce the number of rotational conformations available for thermal motion. It is this reduction in the number of available states that causes the entropy to decrease. As the chain continues to straighten, all of the isoprene units in the chain are eventually forced into extended conformations and the chain is considered to be ‘taut’. A force constant for chain extension can be estimated from the resulting change in free energy associated with this entropy change.[6] As with regime Ia, the force model for this regime is linear and proportional to the temperature divided by the chain tortuosity.
High chain extension regime, II
When all of the isoprene units in a network chain have been forced to reside in just a few extended rotational conformations, the chain becomes taut. It may be regarded as sensibly straight, except for the zigzag path that the C-C bonds make along the chain contour. However, further extension is still possible by bond distortions, e.g., bond angle increases, bond stretches and dihedral angle rotations. These forces are spring-like and are not associated with entropy changes. A taut chain can be extended by only about 40%. At this point the force along the chain is sufficient to mechanically rupture the C-C covalent bond. This tensile force limit has been calculated[7] via quantum chemistry simulations and it is approximately 7 nN, about a factor of a thousand greater than the entropic chain forces at low strain. The angles between adjacent backbone C-C bonds in an isoprene unit vary between about 115-120 degrees and the forces associated with maintaining these angles are quite large, so within each unit, the chain backbone always follows a zigzag path, even at bond rupture. This mechanism accounts for the steep upturn in the elastic stress, observed at high strains (Fig. 1).
Network morphology
Although the network is completely described by only two parameters (the number of network nodes per unit volume and the statistical de-correlation length of the polymer, the Kuhn length), the way in which the chains are connected is actually quite complicated. There is a wide variation in the lengths of the chains and most of them are not connected to the nearest neighbor network node. Both the chain length and its end-to-end distance are described by probability distributions. The term ‘morphology’ refers to this complexity. If the cross-linking agent is thoroughly mixed, there is an equal probability for any isoprene unit to become a network node. For dicumyl peroxide, the cross linking efficiency in natural rubber is unity,[14] but this is not the case for sulfur.[15] The initial morphology of the network is dictated by two random processes: the probability for a cross-link to occur at any isoprene unit and the Markov random walk nature of a chain conformation.[8][9] The probability distribution function for how far one end of a chain end can ‘wander’ from the other is generated by a Markov sequence.[16] This conditional probability density function relates the chain length in units of the Kuhn length to the end-to-end distance :



-
(1)

The probability that any isoprene unit becomes part of a cross-link node is proportional to the ratio of the concentrations of the cross-linker molecules (e.g., dicumyl-peroxide) to the isoprene units:

The factor of two comes about because two isoprene units (one from each chain) participate in the cross-link. The probability for finding a chain containing isoprene units is given by:

-
(3)

where . The equation can be understood as simply the probability that an isoprene unit is NOT a cross-link (1-px) in N-1 successive units along a chain. Since P(N) decreases with N, shorter chains are more probable than longer ones. Note that the number of statistically independent backbone segments is not the same as the number of isoprene units. For natural rubber networks, the Kuhn length contains about 2.2 isoprene units, so . It is the product of equations (1) and (3) (the joint probability distribution) that relates the network chain length () and end-to-end distance () between its terminating cross-link nodes:


-
(4)

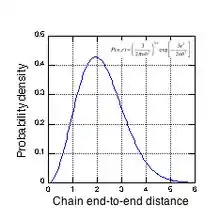
The complex morphology of a natural rubber network can be seen in Fig. 3, which shows the probability density vs. end-to-end distance (in units of mean node spacing) for an ‘average’ chain. For the common experimental cross-link density of 4x1019 cm−3, an average chain contains about 116 isoprene units (52 Kuhn lengths) and has a contour length of about 50 nm. Fig. 3 shows that a significant fraction of chains span several node spacings, i.e., the chain ends overlap other network chains. Natural rubber, cross-linked with dicumyl peroxide, has tetra-functional cross-links, i.e. each cross-link node has 4 network chains emanating from it. Depending on their initial tortuosity and the orientation of their endpoints with respect to the strain axis, each chain associated with an active cross-link node can have a different elastic force constant as it resists the applied strain. To preserve force equilibrium (zero net force) on each cross-link node, a node may be forced to move in tandem with the chain having the highest force constant for chain extension. It is this complex node motion, arising from the random nature of the network morphology, that makes the study of the mechanical properties of rubber networks so difficult. As the network is strained, paths composed of these more extended chains emerge that span the entire sample, and it is these paths that carry most of the stress at high strains.
Numerical network simulation model
To calculate the elastic response of a rubber sample, the three chain force models (regimes Ia, Ib and II) and the network morphology must be combined in a micro-mechanical network model.[10][11][12] Using the joint probability distribution in equation (4) and the force extension models, it is possible to devise numerical algorithms to both construct a faithful representative volume element of a network and to simulate the resulting mechanical stress as it is subjected to strain. An iterative relaxation algorithm is used to maintain approximate force equilibrium at each network node as strain is imposed. When the force constant obtained for kinks having 2 or 3 isoprene units (approximately one Kuhn length) is used in numerical simulations, the predicted stress is found to be consistent with experiments. The results of such a calculation[15] are shown in Fig. 1 (dashed red line) for sulfur cross-linked natural rubber and compared with experimental data[17] (solid blue line). These simulations also predict a steep upturn in the stress as network chains become taut and, ultimately, material failure due to bond rupture. In the case of sulfur cross-linked natural rubber, the S-S bonds in the cross-link are much weaker than the C-C bonds on the chain backbone and are the network failure points. The plateau in the simulated stress, starting at a strain of about 7, is the limiting value for the network. Stresses greater than about 7 MPa cannot be supported and the network fails. Near this stress limit, the simulations predict[12] that less than 10% of the chains are taut, i.e. in the high chain extension regime and less than 0.1% of the chains have ruptured. While the very low rupture fraction may seem surprising, it is not inconsistent with our experience of stretching a rubber band until it breaks. The elastic response of the rubber after breaking is not noticeably different from the original.
Experiments
Variation of tensile stress with temperature
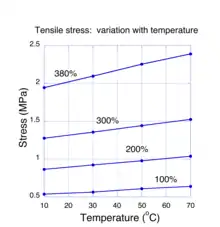
For molecular systems in thermal equilibrium, the addition of energy. e. g. by mechanical work, can cause a change in entropy. This is known from the theories of thermodynamics and statistical mechanics. Specifically, both theories assert that the change in energy must be proportional to the entropy change times the absolute temperature. This rule is only valid so long as the energy is restricted to thermal states of molecules. If a rubber sample is stretched far enough, energy may reside in non-thermal states such as the distortion of chemical bonds and the rule doesn't apply. At low to moderate strains, theory predicts that the required stretching force is due to a change in entropy in the network chains. If this is correct, then we expect that the force necessary to stretch a sample to some value of strain should be proportional to the temperature of the sample. Measurements showing how the tensile stress in a stretched rubber sample varies with temperature are shown in Fig. 4. In these experiments,[18] the strain of a stretched rubber sample was held fixed as the temperature was varied between 10 and 70 degrees Celsius. For each value of fixed strain, it is seen that the tensile stress varied linearly (to within experimental error). These experiments provide the most compelling evidence that entropy changes are the fundamental mechanism for rubber elasticity. The positive linear behavior of the stress with temperature sometimes leads to the mistaken notion that rubber has a negative coefficient of thermal expansion, i.e. the length of a sample shrinks when heated. Experiments[19] have shown conclusively that, like almost all other materials, the coefficient of thermal expansion natural rubber is positive.
Snap-back velocity
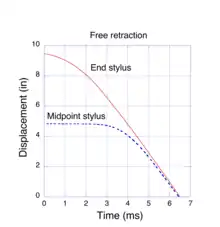
When we stretch a piece of rubber, e.g. a rubber band, we notice that it deforms uniformly, lengthwise. Every element along its length experiences the same extension factor as the entire sample. If we release one end, the sample snaps back to its original length very rapidly, too fast for our eye to resolve the process. Our intuitive expectation is that it returns to its original length in the same manner as when it was stretched, i. e. uniformly. However, this is not what happens. Experimental observations by Mrowca et al.[20] show a surprising behavior. To capture the extremely fast retraction dynamics, they utilized a clever experimental method devised by Exner and Stefan[21] in 1874, well before high-speed electronic measuring devices were invented. Their method consisted of a rapidly rotating glass cylinder which, after being coated with lamp black, was placed next to the stretched rubber sample. Styli, attached to the mid-point and free end of the rubber sample, were held in contact with the glass cylinder. Then, as the free end of the rubber snapped back, the styli traced out helical paths in the lamp black coating of the rotating cylinder. By adjusting the rotation speed of the cylinder, they could record the position of the styli in less than one complete rotation. The trajectories were transferred to a graph by rolling the cylinder on a piece of damp blotter paper. The mark left by a stylus appeared as a white line (no lamp black) on the paper. Their data, plotted as the graph in Fig. 5, shows the position of end and midpoint styli as the sample rapidly retracts to its original length. The sample was initially stretched 9.5” beyond its unstrained length and then released. The styli returned to their original positions (displacement of 0”) in a little over 6 ms. The linear behavior of the displacement vs. time indicates that, after a brief acceleration, both the end and the midpoint of the sample snapped back at a constant velocity of about 50 m/s or 112 mph. However, the midpoint stylus did not start to move until about 3 ms after the end was released. Evidently, the retraction process travels as a wave, starting at the free end. At high extensions some of the energy stored in the stretched network chain is due to a change in its entropy, but most of the energy is stored in bond distortions (regime II, above) which do not involve an entropy change. If one assumes that all of the stored energy is converted to kinetic energy, the retraction velocity may be calculated directly from the familiar conservation equation E= ½ mv2. Numerical simulations,[11] based on the Molecular Kink paradigm, predict velocities consistent with this experiment.
Historical approaches to elasticity theory
Eugene Guth and Hubert M. James proposed the entropic origins of rubber elasticity in 1941.[22]
Thermodynamics
Temperature affects the elasticity of elastomers in an unusual way. When the elastomer is assumed to be in a stretched state, heating causes them to contract. Vice versa, cooling can cause expansion.[23] This can be observed with an ordinary rubber band. Stretching a rubber band will cause it to release heat (press it against your lips), while releasing it after it has been stretched will lead it to absorb heat, causing its surroundings to become cooler. This phenomenon can be explained with the Gibbs free energy. Rearranging ΔG=ΔH−TΔS, where G is the free energy, H is the enthalpy, and S is the entropy, we get TΔS=ΔH−ΔG. Since stretching is nonspontaneous, as it requires external work, TΔS must be negative. Since T is always positive (it can never reach absolute zero), the ΔS must be negative, implying that the rubber in its natural state is more entangled (with more microstates) than when it is under tension. Thus, when the tension is removed, the reaction is spontaneous, leading ΔG to be negative. Consequently, the cooling effect must result in a positive ΔH, so ΔS will be positive there.[24][25]
The result is that an elastomer behaves somewhat like an ideal monatomic gas, inasmuch as (to good approximation) elastic polymers do not store any potential energy in stretched chemical bonds or elastic work done in stretching molecules, when work is done upon them. Instead, all work done on the rubber is "released" (not stored) and appears immediately in the polymer as thermal energy. In the same way, all work that the elastic does on the surroundings results in the disappearance of thermal energy in order to do the work (the elastic band grows cooler, like an expanding gas). This last phenomenon is the critical clue that the ability of an elastomer to do work depends (as with an ideal gas) only on entropy-change considerations, and not on any stored (i.e., potential) energy within the polymer bonds. Instead, the energy to do work comes entirely from thermal energy, and (as in the case of an expanding ideal gas) only the positive entropy change of the polymer allows its internal thermal energy to be converted efficiently (100% in theory) into work.
Polymer chain theories
Invoking the theory of rubber elasticity, one considers a polymer chain in a cross-linked network as an entropic spring. When the chain is stretched, the entropy is reduced by a large margin because there are fewer conformations available.[26] Therefore, there is a restoring force, which causes the polymer chain to return to its equilibrium or unstretched state, such as a high entropy random coil configuration, once the external force is removed. This is the reason why rubber bands return to their original state. Two common models for rubber elasticity are the freely-jointed chain model and the worm-like chain model.
Freely-jointed chain model
The freely joined chain, also called an ideal chain, follows the random walk model. Microscopically, the 3-D random walk of a polymer chain assumes the overall end-to-end distance is expressed in terms of the x, y and z directions:
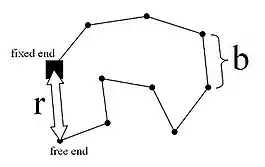

In the model, is the length of a rigid segment, is the number of segments of length , is the distance between the fixed and free ends, and is the "contour length" or . Above the glass transition temperature, the polymer chain oscillates and changes over time. The probability distribution of the chain is the product of the probability distributions of the individual components, given by the following Gaussian distribution:







Therefore, the ensemble average end-to-end distance is simply the standard integral of the probability distribution over all space. Note that the movement could be backwards or forwards, so the net average will be zero. However, one can use the root mean square as a useful measure of the distance.



The Flory theory of rubber elasticity has pointed out the rubber elasticity has primarily entropic origins. By using the following basic equations for Helmholtz free energy and its discussion about entropy, the force generated from the deformation of a rubber chain from its original un-stretched conformation can be derived. The is the number of conformations of the polymer chain. Since the deformation does not involve enthalpy change, the change in free energy can just be calculated as the change in entropy. It can be observed that the force equation resembles the behavior of a spring and follows Hooke's law:, where F is the force, k is the spring constant and x is the distance. Usually, neo-Hookean model can be used on cross-linked polymers to predict their stress-strain relations:







Note that the elastic coefficient is temperature dependent. If we increase the rubber temperature, the elastic coefficient also rises. This is the reason why rubber under constant stress shrinks when its temperature increases.

We can further expand the Flory theory into a macroscopic view, where bulk rubber material is discussed. Assume the original dimension of the rubber material is , and , a deformed shape can then be expressed by applying an individual extension ratio to the length (, , ). So microscopically, the deformed polymer chain can also be expressed with the extension ratio: , , . The free energy change due to deformation can then be expressed as follows:











Assume that the rubber is cross-linked and isotropic, the random walk model gives , and are distributed according to a normal distribution. Therefore, they are equal in space, and all of them are 1/3 of the overall end-to-end distance of the chain: . Plugging in the change of free energy equation above, it is easy to get:





The free energy change per volume is just:

where is the number of strands in network, the subscript means "deformation", , which is the number density per volume of polymer chains, which is the ratio between the end-to-end distance of the chain and the theoretical distance that obey random walk statistics. If we assume incompressibility, the product of extension ratios is 1, implying no change in the volume:.





Case study: Uniaxial deformation:
In a uniaxial deformed rubber, because we assume . So the previous free energy per volume equation is:


The engineering stress (by definition) is the first derivative of the energy in terms of the extension ratio, which is equivalent to the concept of strain:

and the Young's Modulus is defined as derivative of the stress with respect to strain, which measures the stiffness of the rubber in laboratory experiments.


where , is the mass density of the chain, is the number average molecular weight of a network strand between crosslinks. Here, this type of analysis links the thermodynamic theory of rubber elasticity to experimentally measurable parameters. In addition, it gives in sights into the cross-linking condition of the materials.



Worm-like chain model
The worm-like chain model (WLC) takes the energy required to bend a molecule into account. The variables are the same except that , the persistence length, replaces . Then, the force follows this equation:


Therefore, when there is no distance between chain ends (r=0), the force required to do so is zero, and to fully extend the polymer chain (), an infinite force is required, which is intuitive. Graphically, the force begins at the origin and initially increases linearly with . The force then plateaus but eventually increases again and approaches infinity as the chain length approaches .


References
- Proc. Lit. and Phil. Soc., Manchester, 2d ser., 1, 288 (1805)
- Lord Kelvin, Quarterly J. Math., 1, 57 (1857)
- Joule JP. On thermodynamic properties of solids. Phil Trans R Soc Lond. 1859;149:91–131.
- D. E. Hanson and J. L. Barber, Contemporary Physics 56 (3), 319-337 (2015), LAPR-2015-022971
- D. E. Hanson and R. L. Martin, Journal of Chemical Physics 133, 084903 (084908 pp.) (2010)
- D. E. Hanson, J. L. Barber and G. Subramanian, Journal of Chemical Physics 139 (2013), LAPR-2014-018991
- D. E. Hanson and R. L. Martin, The Journal of Chemical Physics 130, 064903 (2009), LAPR-2009-006764
- P. Flory, N. Rabjohn and M. Shaffer, Journal of Polymer Science 4, 435-455 (1949)
- D. E. Hanson, Journal of Chemical Physics 134, 064906 (064906 pp.) (2011)
- D. E. Hanson, Polymer 45 (3), 1058-1062 (2004)
- D. E. Hanson, Journal of Chemical Physics 131, 224904 (224905 pp.) (2009)
- D. E. Hanson and J. L. Barber, Modelling and Simulation in Materials Science and Engineering 21 (2013), LAPR-2013-017962
- J. P. Joule, Phil. Trans. R. Soc. London 149, 91–131 (1859)
- L.D. Loan, Pure Appl. Chem. 30 (1972)
- D. E. Hanson and J. L. Barber, Phys. Chem. Chem. Phys. 20, 8460 (2018), LAPR-2018-029488
- A. A. Markov, Izv. Peterb. Akad. 4 (1), 61-80 (1907)
- L. R. G. Treloar, Trans. Faraday Soc., 40, 0059 (1944)
- R. L. Anthony, R. H Caston and Eugene Guth, J. Phys. Chem. 46, 8, (1942 )
- L. A. Wood and G. Martin, Journal of Research of the National Bureau of Standards-A. Physics and Chemistry Vol 68A, No. 3 (1964).
- B. A. Mrowca, S. L. Dart and E. Guth, Physical Review 66, 30 (1944).
- G. S. Whitby, "Plantation Rubber and the Testing of Rubber", Longmans and Green, London, 1920. p 461
- Guth, Eugene; James, Hubert M. (May 1941). "Elastic and Thermoelastic Properties of Rubber like Materials". Ind. Eng. Chem. 33 (5): 624–629. doi:10.1021/ie50377a017.
- "Thermodynamics of a Rubber Band", American Journal of Physics, 31 (5): 397–397, May 1963, Bibcode:1963AmJPh..31..397T, doi:10.1119/1.1969535
- Rubber Bands and Heat, http://scifun.chem.wisc.edu/HomeExpts/rubberband.html%5B%5D, citing Shakhashiri (1983)
- Shakhashiri, Bassam Z. (1983), Chemical Demonstrations: A Handbook for Teachers of Chemistry, 1, Madison, WI: The University of Wisconsin Press, ISBN 978-0-299-08890-3
- L.R.G. Treloar (1975), Physics of Rubber Elasticity, Oxford University Press, ISBN 9780198570271