Rotor syndrome
Rotor syndrome (also known as Rotor type hyperbilirubinemia)[2] is a rare cause of mixed direct (conjugated) and indirect (unconjugated) hyperbilirubinemia, relatively benign, autosomal recessive[3] bilirubin disorder characterized by non-hemolytic jaundice due to the chronic elevation of predominantly conjugated bilirubin.[2]
| Rotor syndrome | |
|---|---|
| Other names | Rotor type hyperbilirubinemia[1] |
 | |
| Bilirubin | |
| Specialty | Pediatrics, hepatology |
Rotor type hyperbilirubinemia is a distinct yet similar disorder to Dubin–Johnson syndrome[1] – both diseases cause an increase in conjugated bilirubin. Whereas rotor syndrome differs in that it is a result of impaired hepatocellular storage of conjugated bilirubin that leaks into plasma causing hyperbilirubinemia.[2]
Signs and symptoms
Rotor syndrome has many features in common with Dubin–Johnson syndrome, an exception being that the liver cells are not pigmented. The main symptom is a non-itching jaundice. There is a rise in bilirubin in the patient's serum, mainly of the conjugated type.
It can be differentiated from Dubin–Johnson syndrome in the following ways:[4]
| Rotor syndrome | Dubin–Johnson syndrome | |
| appearance of liver | normal histology and appearance | liver has black pigmentation |
| gallbladder visualization | gallbladder can be visualized by oral cholecystogram | gallbladder cannot be visualized |
| total urine coproporphyrin content | high with <70% being isomer 1 | normal with >80% being isomer 1 (normal urine contains more of isomer 3 than isomer 1) |
It has been suggested that Rotor syndrome may exacerbate toxic side effects of the medication irinotecan.[5]
Pathophysiology
Rotor syndrome is caused by mutations in two proteins responsible for transporting bilirubin and other compounds from the blood to the liver to be metabolized and cleared from the body.[2]
Coproporphyrin I, a major coproporphyrin isomer in bile, is transported from the hepatocyte back into the circulation and is excreted in the urine. Thus, urine coproporphyrin is elevated in Rotor syndrome.[2]
Cholescintigraphy using sulfobromophthalein (BSP) have shown that the transport capacity of dye into bile is reduced by less than 50%, and the storage capacity in the hepatocytes is decreased more than 5-fold compared with normal values in this disease.[2]
Genetics
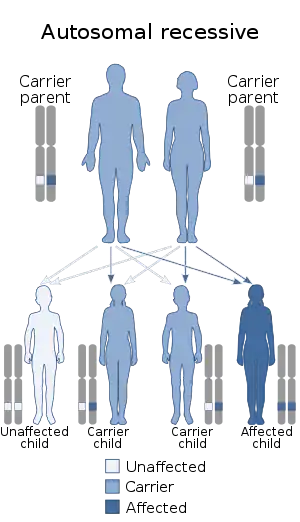
Rotor syndrome is inherited in an autosomal recessive manner.[3] The SLCO1B1 and SLCO1B3 genes are involved in Rotor syndrome.[6] Mutations in both genes are required for the condition to occur. The SLCO1B1 and SLCO1B3 genes provide instructions for making similar proteins, called organic anion transporting polypeptide 1B1 (OATP1B1) and organic anion transporting polypeptide 1B3 (OATP1B3), respectively. Both proteins are found in liver cells; they transport bilirubin and other compounds from the blood into the liver so that they can be cleared from the body. In the liver, bilirubin is dissolved in a digestive fluid called bile and then excreted from the body. The SLCO1B1 and SLCO1B3 gene mutations that cause Rotor syndrome lead to abnormally short, nonfunctional OATP1B1 and OATP1B3 proteins or an absence of these proteins. Without the function of either transport protein, bilirubin is less efficiently taken up by the liver and removed from the body. The buildup of this substance leads to jaundice in people with Rotor syndrome.[7]
Diagnosis
Increased conjugated hyperbilirubinemia is the hallmark for diagnosing Rotor syndrome. There is no distinct black pigmentation of the liver as seen in a similar, Dubin-Johnson Syndrome. Genes, SLCO1B1 and SLCO1B3 that result in complete functional deficiencies of both protein products (OATP1B1 and OATP1B3, respectively), are also present.
Rotor syndrome is largely a diagnosis of exclusion.[2] Serological abnormalities in Rotor syndrome only include elevated total serum bilirubin (typically elevated between 2 to 5 mg/dL but may be as high as 20 mg/dL).[2]
Most of the time, alanine aminotransferase, aspartate aminotransferase, gamma-glutamyl transferase, and alkaline phosphatase levels are normal, but mild elevations can be seen.[2] If any of these lab values are markedly elevated, investigation for other, more serious conditions is warranted.[2]
Imaging studies cannot diagnose Rotor syndrome but can help rule out other diseases that cause hyperbilirubinemia.[2] For example, ultrasound of the liver and the biliary tree can help investigate the causes of extra-hepatic biliary obstruction.[2] The gallbladder is visualized on oral cholecystography in Rotor syndrome while it is not visualized in Dubin Johnson syndrome.[2] Ultimately, the best method of diagnosing the disease is the analysis of urine coproporphyrin excretion.[2] The total urine coproporphyrin excretion in Rotor syndrome has a 2- to 5-fold elevation, with 65% constituting coproporphyrin I.[2]
Treatment
To treat the jaundice, phenobarbital is normally used.
Rotor syndrome is a benign disease requiring no treatment.[2] Jaundice is a lifelong finding, but the disease is not associated with morbidity or mortality, and life expectancy is not affected.[2] Most individuals with Rotor syndrome are born to consanguineous couples and its diagnosis may coincidently identify consanguinity.[2] Distinguishing Rotor syndrome from other more serious disorders is important to avoid unnecessary workup and interventions.[2] It is also critical to reassure and calm patients or family members of patients with Rotors syndrome that the condition is benign.[2]
Eponym
Rotor syndrome is named after the Filipino internist Arturo Belleza Rotor (1907–1988).[8]
References
- Online Mendelian Inheritance in Man (OMIM): 237450
- Kumar, A; Mehta, D (2020), "article-36575", Rotor Syndrome, This book is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, a link is provided to the Creative Commons license, and any changes made are indicated., Treasure Island (FL): StatPearls Publishing, PMID 30335339, retrieved 2020-07-17
- Wolkoff AW, Wolpert E, Pascasio FN, Arias IM (February 1976). "Rotor's syndrome. A distinct inheritable pathophysiologic entity". The American Journal of Medicine. 60 (2): 173–179. doi:10.1016/0002-9343(76)90426-5. PMID 766621.
- Robert Wyllie; Jeffrey S. Hyams (2010-11-29). Pediatric Gastrointestinal and Liver Disease E-Book. Elsevier Health Sciences. pp. 186–. ISBN 978-1-4377-3566-6.
- Iusuf, D., Ludwig, M., Elbatsh, A., van Esch, A., van de Steeg, E., & Wagenaar, E. et al. (2013). OATP1A/1B Transporters Affect Irinotecan and SN-38 Pharmacokinetics and Carboxylesterase Expression in Knockout and Humanized Transgenic Mice. Molecular Cancer Therapeutics, 13(2), 492-503. https://dx.doi.org/10.1158/1535-7163.mct-13-0541\
- van de Steeg E, Stránecký V, Hartmannová H, Nosková L, Hřebíček M, Wagenaar E, van Esch A, de Waart DR, Oude Elferink RP, Kenworthy KE, Sticová E, al-Edreesi M, Knisely AS, Kmoch S, Jirsa M, Schinkel AH (2012). "Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver". The Journal of Clinical Investigation. 122 (2): 519–28. doi:10.1172/JCI59526. PMC 3266790. PMID 22232210.
- "Rotor Syndrome". NIH. U.S. Department of Health & Human Services.
- synd/2296 at Who Named It?
External links
- Hyperbilirubinemia, Conjugated at eMedicine
- Rotor syndrome at NIH's Office of Rare Diseases
- Mentioned in MedlinePlus Encyclopedia: Jaundice – yellow skin
| Classification | |
|---|---|
| External resources |