Pseudo-Hurler polydystrophy
Pseudo-Hurler polydystrophy, also referred to as mucolipidosis III (ML III), is a lysosomal storage disease closely related to I-cell disease (ML II).[2] This disorder is called Pseudo-Hurler because it resembles a mild form of Hurler syndrome, one of the mucopolysaccharide (MPS) diseases.
| Pseudo-Hurler polydystrophy | |
|---|---|
| Other names | Mucolipidosis III alpha/beta [1] |
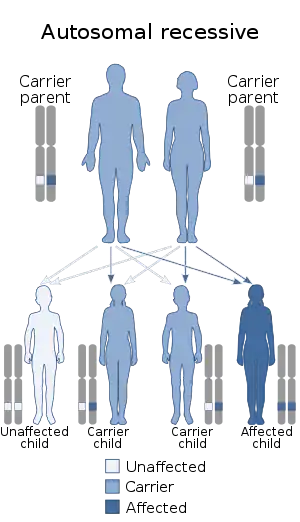 | |
| Pseudo-Hurler polydystrophy is inherited in an autosomal recessive manner | |
| Specialty | Medical genetics |
Signs and symptoms
Symptoms of ML III are often not noticed until the child is 3–5 years of age. Patients with ML III are generally of normal intelligence (trait) or have only mild mental retardation. These patients usually have skeletal abnormalities, coarse facial features, short height, corneal clouding, carpal tunnel syndrome, aortic valve disease and mild enlargement of organs. Some children with severe forms of this disease do not live beyond childhood. However, there is a great variability among patients - there are diagnosed individuals with ML III living in their sixties.
Pathophysiology
As in Mucolipidosis II, Mucolipidosis III results from genetic defects in GlcNAc phosphotransferase (N-acetylglucosamine-1-phosphotransferase). However, ML III produces less severe symptoms and progresses more slowly, probably because the defect in GlcNAc phosphotranspherase lies in its protein recognition domain.[3] Therefore, the catalytic domain retains some of its activity, resulting in a smaller accumulation of carbohydrates, lipids, and proteins in the inclusion bodies.
Treatment
There is no cure for Pseudo-Hurler Polydystrophy/Mucolipidosis IIIA. Treatment is limited to controlling or reducing symptoms associated with this disorder. Physio-therapy, particularly hydrotherapy has proven effective at relieving muscle stiffness and increasing mobility. The use of crutches, a wheelchair or scooters are treatment options as the metabolic bone disease progresses. The insertion of rods in the spine to stabilize the vulnerable areas can treat scoliosis. Heart valve replacement surgery may be necessary as this disorder progresses, Enzyme replacement therapy has been suggested as a potential treatment
See also
References
- "Mucolipidosis III alpha/beta | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 11 April 2019.
- Bargal R, Zeigler M, Abu-Libdeh B, et al. (August 2006). "When Mucolipidosis III meets Mucolipidosis II: GNPTA gene mutations in 24 patients". Mol. Genet. Metab. 88 (4): 359–63. doi:10.1016/j.ymgme.2006.03.003. PMID 16630736.
- Murray, R, Granner, D, and Rodwell, V. (2006). Harper's Illustrated Biochemistry. 27th ed. New York: Lange Medical Books/McGraw-Hill.
External links
| Classification |
|---|
- Mucolipidosis type 3 A at NIH's Office of Rare Diseases
- mucolipidoses at NINDS - original text of article derived from detail sheet available here