Leber's hereditary optic neuropathy
Leber's hereditary optic neuropathy (LHON) is a mitochondrially inherited (transmitted from mother to offspring) degeneration of retinal ganglion cells (RGCs) and their axons that leads to an acute or subacute loss of central vision; this affects predominantly young adult males. LHON is only transmitted through the mother, as it is primarily due to mutations in the mitochondrial (not nuclear) genome, and only the egg contributes mitochondria to the embryo. LHON is usually due to one of three pathogenic mitochondrial DNA (mtDNA) point mutations. These mutations are at nucleotide positions 11778 G to A, 3460 G to A and 14484 T to C, respectively in the ND4, ND1 and ND6 subunit genes of complex I of the oxidative phosphorylation chain in mitochondria. Men cannot pass on the disease to their offspring.[1]
| Leber's hereditary optic neuropathy | |
|---|---|
| Other names | Leber hereditary optic atrophy |
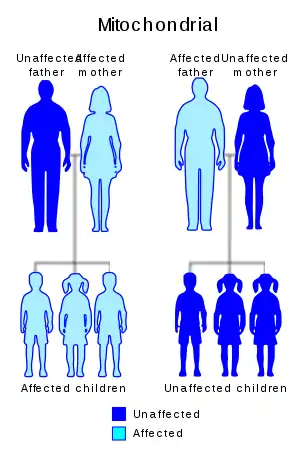 | |
| Leber’s hereditary optic neuropathy has a mitochondrial inheritance pattern. | |
| Specialty | Ophthalmology |
| Frequency | 1:30,000 to 1:50,000 |
Signs and symptoms
Clinically, there is an acute onset of visual loss, first in one eye, and then a few weeks to months later in the other. Onset is usually young adulthood, but age range at onset from 7-75 is reported. The age of onset is slightly higher in females (range 19–55 years: mean 31.3 years) than males (range 15–53 years: mean 24.3). The male to female ratio varies between mutations: 3:1 for 3460 G>A, 6:1 for 11778 G>A and 8:1 for 14484 T>C.
This typically evolves to very severe optic atrophy and a permanent decrease of visual acuity. Both eyes become affected either simultaneously (25% of cases) or sequentially (75% of cases) with a median inter-eye delay of 8 weeks. Rarely only one eye may be affected. In the acute stage, lasting a few weeks, the affected eye demonstrates an oedematous appearance of the nerve fiber layer especially in the arcuate bundles and enlarged or telangiectatic and tortuous peripapillary vessels (microangiopathy). The main features are seen on fundus examination, just before or subsequent to the onset of visual loss. A pupillary defect may be visible in the acute stage as well. Examination reveals decreased visual acuity, loss of color vision and a cecocentral scotoma on visual field examination.
LHON with demyelinating lesions or LHON Plus
"LHON Plus" is a name given to a rare variant of the disorder with eye disease together with other conditions.[2]
The symptoms of this higher form of the disease include loss of the brain's ability to control the movement of muscles, tremors, and cardiac arrhythmia.[3] Many cases of LHON plus have been comparable to multiple sclerosis because of the lack of muscular control[4] and because of the presence of demyelinating lesions in the CNS. It is therefore a subtype of MS according to McDonalds definition.[5]
Genetics
Leber hereditary optic neuropathy is a condition related to changes in mitochondrial DNA. Although most DNA is packaged in chromosomes within the nucleus, mitochondria have a distinct mitochondrial genome composed of mtDNA.
Mutations in the MT-ND1, MT-ND4, MT-ND4L, and MT-ND6 genes cause Leber hereditary optic neuropathy.[6] These genes code for the NADH dehydrogenase protein involved in the normal mitochondrial function of oxidative phosphorylation. Oxidative phosphorylation uses a series of four large multienzyme complexes, which are all embedded in the inner mitochondrial membrane to convert oxygen and simple sugars to energy. Mutations in any of the genes disrupt this process to cause a variety of syndromes depending on the type of mutation and other factors. It remains unclear how these genetic changes cause the death of cells in the optic nerve and lead to the specific features of Leber hereditary optic neuropathy.
Pathophysiology
The eye pathology is limited to the retinal ganglion cell layer especially the maculopapillary bundle. Degeneration is evident from the retinal ganglion cell bodies to the axonal pathways leading to the lateral geniculate nuclei. Experimental evidence reveals impaired glutamate transport and increased reactive oxygen species (ROS) causing apoptosis of retinal ganglion cells. Also, experiments suggest that normal non LHON affected retinal ganglion cells produce less of the potent superoxide radical than other normal central nervous system neurons.[7] Viral vector experiments which augment superoxide dismutase 2 in LHON cybrids[8] or LHON animal models or use of exogenous glutathione in LHON cybrids[9] have been shown to rescue LHON affected retinal ganglion cells from apoptotic death. These experiments may in part explain the death of LHON affected retinal ganglion cells in preference to other central nervous system neurons which also carry LHON affected mitochondria.
Diagnosis
Without a known family history of LHON the diagnosis usually requires a neuro-ophthalmological evaluation and blood testing for mitochondrial DNA assessment.[10] It is important to exclude other possible causes of vision loss and important associated syndromes such as heart electrical conduction system abnormalities.
Treatment
The prognosis for those affected left untreated is almost always that of continued significant visual loss in both eyes. Regular corrected visual acuity and perimetry checks are advised for follow up of affected individuals. There is beneficial treatment available for some cases of this disease especially for early onset disease.[11] Also, experimental treatment protocols are in progress.[12] Genetic counselling should be offered. Health and lifestyle choices should be reassessed particularly in light of toxic and nutritional theories of gene expression. Vision aids assistance and work rehabilitation should be used to assist in maintaining employment.
For those who are carriers of a LHON mutation, preclinical markers may be used to monitor progress.[13] For example, fundus photography can monitor nerve fiber layer swelling. Optical coherence tomography can be used for more detailed study of retinal nerve fiber layer thickness. Red green color vision testing may detect losses. Contrast sensitivity may be diminished. There could be an abnormal electroretinogram or visual evoked potentials. Neuron-specific enolase and axonal heavy chain neurofilament blood markers may predict conversion to affected status.
Cyanocobalamin (a form of B12) may also be used.[14]
Avoiding optic nerve toxins is generally advised, especially tobacco and alcohol. Certain prescription drugs are known to be a potential risk, so all drugs should be treated with suspicion and checked before use by those at risk. Ethambutol, in particular, has been implicated as triggering visual loss in carriers of LHON. In fact, toxic and nutritional optic neuropathies may have overlaps with LHON in symptoms, mitochondrial mechanisms of disease and management.[15] Of note, when a patient carrying or suffering from LHON or toxic/nutritional optic neuropathy suffers a hypertensive crisis as a possible complication of the disease process, nitroprusside (trade name: Nipride) should not be used due to increased risk of optic nerve ischemia in response to this anti-hypertensive in particular.[16]
Idebenone[11][17][18] has been shown in a small placebo controlled trial to have modest benefit in about half of patients. People most likely to respond best were those treated early in onset.
α-Tocotrienol-quinone, a vitamin E metabolite, has had some success in small open label trials in reversing early onset vision loss.[12][19]
There are various treatment approaches which have had early trials or are proposed, none yet with convincing evidence of usefulness or safety for treatment or prevention including brimonidine,[20] minocycline,[21] curcumin,[22] glutathione,[9] near infrared light treatment,[23] and viral vector techniques.[8]
"Three person in vitro fertilization" is a proof of concept research technique for preventing mitochondrial disease in developing human fetuses. So far, viable macaque monkeys have been produced. But ethical and knowledge hurdles remain before use of the technique in humans is established.[24]
Idebenone
Idebenone is a short-chain benzoquinone that interacts with the mitochondrial electron transport chain to enhance cellular respiration. When used in individuals with LHON, it is believed to allow electrons to bypass the dysfunctional complex I.[25] Successful treatment using idebenone was initially reported in a small number of patients.[18][26]
Two large-scale studies have demonstrated the benefits of idebenone. The Rescue of Hereditary Optic Disease Outpatient Study (RHODOS) evaluated the effects of idebenone in 85 patients with LHON who had lost vision within the prior five years.[11][27] In this study, the group taking idebenone 900 mg per day for 24 weeks showed a slight improvement in visual acuity compared to the placebo group, though this difference was not statistically significant. Importantly, however, patients taking idebenone were protected from further vision loss, whereas the placebo group had a steady decline in visual acuity. Further, individuals taking idebenone demonstrated preservation of color vision and persistence of the effects of idebenone 30 months after discontinuing therapy.[27][28] A retrospective analysis of 103 LHON patients by Carelli et al. builds upon these results.[29] This study highlighted that 44 subjects who were treated with idebenone within one year of onset of vision loss had better outcomes, and, further, that these improvements with idebenone persisted for years.
Idebenone, combined with avoidance of smoke and limitation of alcohol intake, is the preferred standard treatment protocol for patients affected by LHON.[30] Idebenone doses are prescribed to be taken spaced out throughout the day, rather than all at one time. For example, to achieve a dose of 900 mg per day, patients take 300 mg three times daily with meals. Idebenone is fat soluble, and may be taken with a moderate amount of dietary fat in each meal to promote absorption. It is recommended that patients on idebenone also take vitamin C 500 mg daily to keep idebenone in its reduced form,[30] as it is most active in this state.[31]
Estrogen Replacement Therapy
Estrogens have been shown to have a protective role in the pathogenesis of LHON. Experiments using LHON cybrids have demonstrated that the estrogen receptor localizes to the mitochondria where it directly mediates mitochondrial biogenesis. Estrogens upregulate the antioxidant enzyme superoxide dismutase 2 and mitochondrial DNA synthesis. These experiments helped to explain the mechanism behind the lower penetrance of disease among female carriers.[32][33][34] While additional factors have been theorized, the protective role of estrogens appears to be a significant contributor.
In addition to the experimental evidence, clinical data also points towards the protective role of estrogens. Penetrance among female carriers is substantially lower (between 3 and 8 to 1 male to female ratios depending on the mutation) while average age at onset is significantly higher. Multiple case series of various LHON pedigrees have described female carriers converting after menopause or cessation of hormone replacement therapies.[35][36] Together, these form a shifting paradigm towards considering reduced estrogen states, such as menopause, as potential triggers of visual loss similar to smoking or excessive alcohol consumption.
Hormone replacement therapy (HRT) is emerging as an effective therapeutic target for female mutation carriers. In one recent case study where the affected female converted following cessation of HRT, idebenone, and HRT were given together.[35] Visual acuity improved much faster than is typically expected. The patient’s vision returned to 20/40 and 20/60 from 20/60 and 20/200 in the right and left eyes respectively after only one month and was back normal by 8 months compared to the months to years timeframe seen in most cases. While the balance between risks and benefits of HRT remains controversial, the decision to start HRT requires an individualized approach based on the patient’s context. While not applicable for all post-menopausal women, prophylactic (and therapeutic) HRT should be considered in all female carriers of a known LHON mutation given the substantial risk of vision loss associated with menopause.[33][37][35]
Epidemiology
In Northern European populations about one in 9000 people carry one of the three primary LHON mutations.[38] [39] There is a prevalence of between 1:30,000 to 1:50,000 in Europe.
The LHON ND4 G11778A mutation dominates as the primary mutation in most of the world with 70% of Northern European cases and 90% of Asian cases. Due to a Founder effect, the LHON ND6 T14484C mutation accounts for 86% of LHON cases in Quebec, Canada.[40]
More than 50 percent of males with a mutation and more than 85 percent of females with a mutation never experience vision loss or related medical problems. The particular mutation type may predict the likelihood of penetrance, severity of illness and probability of vision recovery in the affected. As a rule of thumb, a woman who harbors a homoplasmic primary LHON mutation has a ~40% risk of having an affected son and a ~10% risk of having an affected daughter.
Additional factors may determine whether a person develops the signs and symptoms of this disorder. Environmental factors such as smoking and alcohol use may be involved, although studies of these factors have produced conflicting results. Researchers are also investigating whether changes in additional genes, particularly genes on the X chromosome,[41] [42] contribute to the development of signs and symptoms. The degree of heteroplasmy, the percentage of mitochondria which have mutant alleles, may play a role.[43] Patterns of mitochondrial alleles called haplogroup may also affect expression of mutations.[44]
History
This disease was first described by the German ophthalmologist Theodor Leber (1840–1917) in 1871.[45] In this paper Leber described four families in which a number of young men suffered abrupt loss of vision in both eyes either simultaneously or sequentially. This disease was initially thought to be X linked but was subsequently shown to be mitochondrial.[46] The nature of the causative mutation was first identified in 1988 by Wallace et al. who discovered the guanine (G) to adenosine (A) mutation at nucleotide position 11778 in nine families.[47] This mutation converts a highly conserved arginine to histidine at codon 340 in the NADH dehydrogenase subunit 4 of complex I of the mitochondrial respiratory chain. The other two mutations known to cause this condition were identified in 1991 (G to A point mutation at nucleotide position 3460)[48] and 1992 (thymidine (T) to cytosine (C) mutation at nucleotide 14484).[49] These three mutations account for over 95% of cases: the 11778 mutation accounts for 50-70% of cases, the 14484 mutation for 10-15% and the 3460 mutation for 8-25%.
Research
Currently, human clinical trials are underway at GenSight Biologics (ClinicalTrials.gov # NCT02064569) and the University of Miami (ClinicalTrials.gov # NCT02161380) to examine the safety and efficacy of mitochondrial gene therapy in LHON. In these trials, participants affected by LHON with the G11778A mutation will have a virus expressing the functional version of ND4 – the gene mutated in this variant of LHON – injected into one eye. A sham injection will be administered to the other eye for comparison. It is hypothesized that introduction of the viral vector may be able to rescue the function of the mutant gene. Preliminary results have demonstrated tolerability of the injections in a small number of subjects.[50]
Stealth BioTherapeutics is presently investigating the potential use of elamipretide (MTP-131), a mitochondrial protective agent, as a therapy for LHON. Elamipretide helps stabilize cardiolipin [51][52] – an important component of mitochondrial inner membranes – and has been shown to reduce damaging reactive oxygen species in animal models.[53] Clinical trials in LHON patients are planned for the future.
See also
References
- Bandelt HJ, Kong QP, Parson W, Salas A (December 2005). "More evidence for non-maternal inheritance of mitochondrial DNA?". J. Med. Genet. 42 (12): 957–60. doi:10.1136/jmg.2005.033589. PMC 1735965. PMID 15923271.
- Nikoskelainen EK, Marttila RJ, Huoponen K, et al. (August 1995). "Leber's "plus": neurological abnormalities in patients with Leber's hereditary optic neuropathy". J. Neurol. Neurosurg. Psychiatry. 59 (2): 160–4. doi:10.1136/jnnp.59.2.160. PMC 485991. PMID 7629530.
- cardiac arrythmia
- Mayo Clinic: Multiple Sclerosis
- David Bargiela, Patrick F Chinnery, Mitochondria in neuroinflammation – Multiple sclerosis (MS), leber hereditary optic neuropathy (LHON) and LHON-MS, https://doi.org/10.1016/j.neulet.2017.06.051
- Online Mendelian Inheritance in Man (OMIM): LEBER OPTIC ATROPHY - 535000
- Hoegger MJ, Lieven CJ, Levin LA (2008). "Differential production of superoxide by neuronal mitochondria". BMC Neurosci. 9: 4. doi:10.1186/1471-2202-9-4. PMC 2266764. PMID 18182110.
- Qi X, Sun L, Hauswirth WW, Lewin AS, Guy J (February 2007). "Use of mitochondrial antioxidant defenses for rescue of cells with a Leber hereditary optic neuropathy-causing mutation". Arch. Ophthalmol. 125 (2): 268–72. doi:10.1001/archopht.125.2.268. PMID 17296905.
- Ghelli A, Porcelli AM, Zanna C, Martinuzzi A, Carelli V, Rugolo M (February 2008). "Protection against oxidant-induced apoptosis by exogenous glutathione in Leber hereditary optic neuropathy cybrids". Invest. Ophthalmol. Vis. Sci. 49 (2): 671–6. doi:10.1167/iovs.07-0880. PMID 18235013.
- Yu-Wai-Man P, Chinnery PF (June 23, 2016). "Leber Hereditary Optic Neuropathy". NCBI. Genereviews. Retrieved February 25, 2018.
- Klopstock, T.; Yu-Wai-Man, P.; Dimitriadis, K.; Rouleau, J.; Heck, S.; Bailie, M.; Atawan, A.; Chattopadhyay, S.; Schubert, M.; Garip, A.; Kernt, M.; Petraki, D.; Rummey, C.; Leinonen, M.; Metz, G.; Griffiths, P. G.; Meier, T.; Chinnery, P. F. (2011). "A randomized placebo-controlled trial of idebenone in Leber's hereditary optic neuropathy". Brain. 134 (9): 2677–2686. doi:10.1093/brain/awr170. ISSN 0006-8950. PMC 3170530. PMID 21788663.
- Shrader, W. D.; Amagata, A.; Barnes, A.; Enns, G. M.; Hinman, A.; Jankowski, O.; Kheifets, V.; Komatsuzaki, R.; Lee, E.; Mollard, P.; Murase, K.; Sadun, A. A.; Thoolen, M.; Wesson, K.; Miller, G. (2011). "Α-Tocotrienol quinone modulates oxidative stress response and the biochemistry of aging". Bioorganic & Medicinal Chemistry Letters. 21 (12): 3693–3698. doi:10.1016/j.bmcl.2011.04.085. PMID 21600768.
- Sadun AA, Salomao SR, Berezovsky A, et al. (2006). "Subclinical carriers and conversions in Leber hereditary optic neuropathy: A prospective psychophysical study". Trans Am Ophthalmol Soc. 104: 51–61. PMC 1809912. PMID 17471325.
- World Health Organization (2009). Stuart MC, Kouimtzi M, Hill SR (eds.). WHO Model Formulary 2008. World Health Organization. p. 251. hdl:10665/44053. ISBN 9789241547659.
- Carelli V, Ross-Cisneros FN, Sadun AA (January 2004). "Mitochondrial dysfunction as a cause of optic neuropathies". Prog Retin Eye Res. 23 (1): 53–89. doi:10.1016/j.preteyeres.2003.10.003. PMID 14766317.
- Katz, Jason; Patel, Chetan (2006). Parkland Manual of Inpatient Medicine. Dallas, TX: FA Davis. p. 903.
- Clinical Idebenone trial recruiting at Newcastle University UK http://lhon.ncl.ac.uk
- Mashima Y, Kigasawa K, Wakakura M, Oguchi Y (September 2000). "Do idebenone and vitamin therapy shorten the time to achieve visual recovery in Leber hereditary optic neuropathy?". J Neuroophthalmol. 20 (3): 166–70. doi:10.1097/00041327-200020030-00006. PMID 11001192.
- Sadun, A et al. "EPI-743 alters the natural history of progression of Leber hereditary optic neuropathy". AOS meeting. May 2011 Archived 2011-09-04 at the Wayback Machine
- Newman NJ, Biousse V, David R, et al. (September 2005). "Prophylaxis for second eye involvement in leber hereditary optic neuropathy: an open-labeled, nonrandomized multicenter trial of topical brimonidine purite". Am. J. Ophthalmol. 140 (3): 407–15. doi:10.1016/j.ajo.2005.03.058. PMID 16083844.
- Haroon MF, Fatima A, Schöler S, et al. (2007). "Minocycline, a possible neuroprotective agent in Leber's hereditary optic neuropathy (LHON): Studies of cybrid cells bearing 11778 mutation". Neurobiol Dis. 28 (3): 237–50. doi:10.1016/j.nbd.2007.07.021. PMID 17822909.
- Clinical Curcurmin trial recruiting at ClinicalTrials.nlm.nih.gov Archived 2009-02-13 at the Wayback Machine
- Wisconsin near infrared trial Archived 2008-05-15 at the Wayback Machine
- Craven L, Tuppen HA, Greggains GD, Harbottle SJ, Murphy JL, Cree LM, Murdoch AP, Chinnery PF, Taylor RW, Lightowlers RN, Herbert M, Turnbull DM (May 2010). "Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease". Nature. 465 (7294): 82–85. Bibcode:2010Natur.465...82C. doi:10.1038/nature08958. PMC 2875160. PMID 20393463.
- Haefeli RH, Erb M, Gemperli AC, Robay D, Courdier Fruh I, Anklin C, Dallmann R, Gueven N (March 2011). "NQO1-dependent redox cycling of idebenone: effects on cellular redox potential and energy levels". PLOS ONE. 6 (3): e17963. Bibcode:2011PLoSO...617963H. doi:10.1371/journal.pone.0017963. PMC 3069029. PMID 21483849.
- Eng, J.G.; Aggarwal, D.; Sadun, A.A. (April 2009). "Idebenone treatment in patients with Leber hereditary optic neuropathy". Invest Ophthalmol Vis Sci. 50 (13). Retrieved March 22, 2016.
- Klopstock T; Metz G; Yu-Wai-Man P; et al. (2013). "Persistence of the treatment effect of idebenone in Leber's hereditary optic neuropathy". Brain. 136 (2): e230. doi:10.1093/brain/aws279. PMC 3572931. PMID 23388409.
- Rudolph, G.; Dimitriadis, K.; Büchner, B.; Heck, S.; Al-Tamami, J.; Seidensticker, F.; Rummey, C.; Leinonen, M.; Meier, T.; Klopstock, T. (March 2013). "Effects of idebenone on color vision in patients with Leber hereditary optic neuropathy". J Neuroophthalmol. 33 (1): 30–36. doi:10.1097/WNO.0b013e318272c643. PMC 3658961. PMID 23263355.
- Carelli V; La Morgia C; Valentino ML; et al. (September 2011). "Idebenone treatment in Leber's hereditary optic neuropathy". Brain. 134 (Part 9): e188. doi:10.1093/brain/awr180. PMID 21810891.
- Karanjia, R.; Sadun, A.A. (2015). "Advances in therapeutic strategies for Leber's hereditary optic neuropathy". Expert Opinion on Orphan Drugs. 3 (12): 1439–1446. doi:10.1517/21678707.2015.1098531.
- Mordente, A.; Martorana, G.E.; Minotti, G; Giardina, B (January 1998). "Antioxidant properties of 2,3-dimethoxy-5-methyl-6-(10-hydroxydecyl)-1,4-benzoquinone (idebenone)". Chem Res Toxicol. 11 (1): 54–63. doi:10.1021/tx970136j. PMID 9477226.
- Giordano, C.; Iommarini, L; Giordano, L; Maresca, A; Pisano, A; Valentino, M L; Caporali, L; Liguori, R; Deceglie, S; Roberti, M; Fanelli, F; Fracasso, F; Ross-Cisneros, F N; D’Adamo, P; Hudson, G; Pyle, A; Yu-Wai-Man, P; Chinnery, P F; Zeviani, M; Salomao, S R; Berezovsky, A; Belfort Jr, R; Ventura, D F; Moraes, M; Filho, M.M.; Barboni, P; Sadun, F; De Negri, A; Sadun, A.A.; Tancredi, A; Mancini, M; d’Amati, G; Polosa, P L; Cantatore, P; Carelli, V (2013). "Efficient mitochondrial biogenesis drives incomplete penetrance in Leber's hereditary optic neuropathy". Brain. 137 (Pt 2): 335–353. doi:10.1093/brain/awt343. PMC 3914475. PMID 24369379.
- Giordano, C.; Montopoli, M; Perli, E; Orlandi, M; Fantin, M; Ross-Cisneros, F.N. L; Caparrotta, L; Martinuzzi, A; Ragazzi, E; Ghelli, A; Sadun, A.A.; d'Amati, G; Carelli, V (2011). "Oestrogens ameliorate mitochondrial dysfunction in Leber's hereditary optic neuropathy". Brain. 134 (Pt 1): 220–234. doi:10.1093/brain/awq276. PMC 3025718. PMID 20943885.
- Pisano, A.; Preziuso, C; Iommarini, L; Perli, E; Grazioli, P; Campese, A.F.; Maresca, A; Montopoli, M; Masuelli, L; Sadun, A.A.; d'Amati, G; Carelli, V; Ghelli, A.M.; Giordano, C (2015). "Targeting estrogen receptor β as preventive therapeutic strategy for Leber's hereditary optic neuropathy". Human Molecular Genetics. 24 (24): 6921–6931. doi:10.1093/hmg/ddv396. PMID 26410888.
- Fantini, M.; Asanad, S; Karanjia, R; Sadun, A.A. (2019). "Hormone replacement therapy in Leber's hereditary optic neuropathy: Accelerated visual recovery in vivo". Journal of Current Ophthalmology. 31: 102–105. doi:10.1016/j.joco.2018.10.003. PMC 6407313. PMID 30899856.
- Hwang, T.J.; Karanjia, R; Moraes-Filho, M.N.; Gale, J; Show Tran, J.; Chu, E.R.; Salomao, S.R.; Berezovsky, A; Belfort Jr., R; Nunes Moraes, M; Sadun, F; DeNegri, A.M.; La Morgia, C; Barboni, P; Ramos, C.; Chicani, C.F.; Quiros, P.A.; Carelli, V; Sadun, A.A. (2017). "Natural History of Conversion of Leber's Hereditary Optic Neuropathy". Ophthalmology. 124 (6): 843–850. doi:10.1016/j.ophtha.2017.01.002. PMID 28196731.
- Hutchinson, C.V.; Walker, J.A.; Davidson, C (2014). "Oestrogen, ocular function and low-level vision: a review". Journal of Endocrinology. 223 (2): R9–R18. doi:10.1530/JOE-14-0349. PMID 25143633.
- Man PY, Griffiths PG, Brown DT, Howell N, Turnbull DM, Chinnery PF (February 2003). "The Epidemiology of Leber Hereditary Optic Neuropathy in the North East of England". Am. J. Hum. Genet. 72 (2): 333–9. doi:10.1086/346066. PMC 379226. PMID 12518276.
- Puomila A, Hämäläinen P, Kivioja S, et al. (October 2007). "Epidemiology and penetrance of Leber hereditary optic neuropathy in Finland". Eur. J. Hum. Genet. 15 (10): 1079–89. doi:10.1038/sj.ejhg.5201828. PMID 17406640.
- Laberge AM, Jomphe M, Houde L, et al. (2005). "A "Fille du Roy" Introduced the T14484C Leber Hereditary Optic Neuropathy Mutation in French Canadians". Am. J. Hum. Genet. 77 (2): 313–7. doi:10.1086/432491. PMC 1224533. PMID 15954041.
- Hudson G, Carelli V, Horvath R, Zeviani M, Smeets HJ, Chinnery PF (2007). "X-Inactivation patterns in females harboring mtDNA mutations that cause Leber hereditary optic neuropathy". Mol. Vis. 13: 2339–43. PMID 18199976.
- Hudson G, Keers S, Yu Wai Man P, et al. (December 2005). "Identification of an X-Chromosomal Locus and Haplotype Modulating the Phenotype of a Mitochondrial DNA Disorder". Am. J. Hum. Genet. 77 (6): 1086–91. doi:10.1086/498176. PMC 1285165. PMID 16380918.
- Chinnery PF, Andrews RM, Turnbull DM, Howell NN (January 2001). "Leber hereditary optic neuropathy: Does heteroplasmy influence the inheritance and expression of the G11778A mitochondrial DNA mutation?". Am. J. Med. Genet. 98 (3): 235–43. doi:10.1002/1096-8628(20010122)98:3<235::AID-AJMG1086>3.0.CO;2-O. PMID 11169561.
- Hudson G, Carelli V, Spruijt L, et al. (August 2007). "Clinical Expression of Leber Hereditary Optic Neuropathy Is Affected by the Mitochondrial DNA–Haplogroup Background". Am. J. Hum. Genet. 81 (2): 228–33. doi:10.1086/519394. PMC 1950812. PMID 17668373.
- Leber T. Ueber hereditaere und congenital angelegte sehnervenleiden (1871) Graefes Arch Clin Exp Ophthalmol. 17:249–291
- Erickson RP (May 1972). "Leber's optic atrophy, a possible example of maternal inheritance". American Journal of Human Genetics. 24 (3): 348–9. PMC 1762279. PMID 5063796.
- Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ, Nikoskelainen EK (December 1988). "Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy". Science. 242 (4884): 1427–30. Bibcode:1988Sci...242.1427W. doi:10.1126/science.3201231. PMID 3201231.
- Huoponen K, Vilkki J, Aula P, Nikoskelainen EK, Savontaus ML (1991). "A new mtDNA mutation associated with Leber hereditary optic neuroretinopathy". Am J Hum Genet. 48 (6): 1147–1153. PMC 1683111. PMID 1674640.
- Johns DR, Neufeld MJ, Park RD (1992). "An ND-6 mitochondrial DNA mutation associated with Leber hereditary optic neuropathy". Biochem Biophys Res Commun. 187 (3): 1551–1557. doi:10.1016/0006-291x(92)90479-5. PMID 1417830.
- Sahel, J.A.; Uretsky, S.; Combal, J.P.; Galy, A.; Thomasson, N.; Fitoussi, S.; Corral-Debrinsky, M.; Honnet, G.; Vignal, C. (June 2015). "Preliminary safety and tolerability results of intravitreal administration of GS010, a recombinant adeno-associated viral vector serotype 2 (rAAV2/2) containing human wildtype mitochondrial NADH dehydrogenase 4 (ND4) gene in patients with Leber Hereditary Optic Neuropathy (LHON) due to the G11778A ND4 mitochondrial DNA mutation". Invest. Ophthalmol. Vis. Sci. 56 (7): 1088. Retrieved March 22, 2016.
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. (July 2013). "The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin". J Am Soc Nephrol. 24 (8): 1250–61. doi:10.1681/ASN.2012121216. PMC 3736700. PMID 23813215.
- Thomas, D.A.; Stauffer, C.; Zhao, K.; Yang, H.; Sharma, V.K.; Szeto, H.H.; Suthanthiran, M. (January 2007). "Mitochondrial targeting with antioxidant peptide SS-31 prevents mitochondrial depolarization, reduces islet cell apoptosis, increases islet cell yield, and improves posttransplantation function". J Am Soc Nephrol. 18 (1): 213–222. doi:10.1681/asn.2006080825. PMID 17151329. Retrieved March 22, 2016.
- Brown DA; Hale SL; Baines CP; et al. (January 2014). "Reduction of early reperfusion injury with the mitochondria-targeting peptide bendavia". J Cardiovasc Pharmacol Ther. 19 (1): 121–132. doi:10.1177/1074248413508003. PMC 4103197. PMID 24288396.
Further reading
- Leber's hereditary optic neuropathy at NLM Genetics Home Reference
- Kerrison JB, Newman NJ (1997). "Clinical spectrum of Leber's hereditary optic neuropathy" (IFOND reprints). Clin. Neurosci. 4 (5): 295–301. PMID 9292259.
- Carelli V, Ross-Cisneros FN, Sadun AA (January 2004). "Mitochondrial dysfunction as a cause of optic neuropathies". Prog Retin Eye Res. 23 (1): 53–89. doi:10.1016/j.preteyeres.2003.10.003. PMID 14766317.
External links
| Classification |
|---|