Dysplastic nevus syndrome
Dysplastic nevus syndrome, also known as familial atypical multiple mole–melanoma (FAMMM) syndrome, is an inherited cutaneous condition described in certain families, and characterized by unusual nevi and multiple inherited melanomas.[2][3] First described in 1820, the condition is inherited in an autosomal dominant pattern, and caused by mutations in the CDKN2A gene. In addition to melanoma, individuals with the condition are at increased risk for pancreatic cancer.
| Dysplastic nevus syndrome | |
|---|---|
| Other names | Atypical mole syndrome (AMS), familial atypical multiple mole–melanoma (FAMMM) syndrome, familial melanoma syndrome,[1] B-K mole syndrome, atypical mole syndrome |
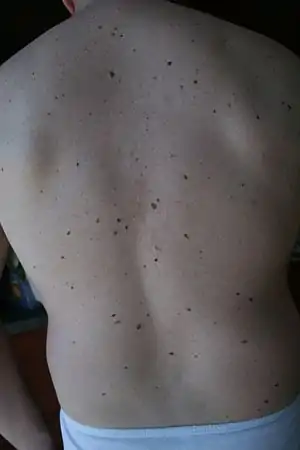 | |
| Large numbers of moles are characteristic features of familial atypical multiple mole–melanoma syndrome. | |
| Specialty | Oncology, medical genetics |
The diagnosis of dysplastic nevus syndrome is based on clinical presentation and family history. Treatment consists of resection of malignant skin lesions (melanoma). Screening for pancreatic cancer may be considered, particularly if there is a family history.
Signs and symptoms
Dysplastic nevus syndrome is characterized by unusual nevi and multiple inherited melanomas.[2]:692[3]
Genetics
The CDKN2A gene is located on chromosome 9p21.3. Two main transcripts, isoforms '1' and '4', each contain three exons and span 7288 and 26740 bp, respectively. They encode proteins of 156 and 173 amino acids; isoform '1' encodes p16(INK4a), while isoform '4' encodes p14(ARF), a protein that is structurally unrelated to p16(INK4) but acts in cell cycle G1 control by stabilizing the tumor suppressor protein p53.[4]
Dysplastic nevus syndrome is inherited in an autosomal dominant manner. The penetrance for melanoma in kindreds with CDKN2A mutations is estimated at 58% to 92% by 80 years of age and varies with geography. The penetrance in CDKN2A mutation carriers for pancreatic cancer has been estimated to be 17% by 75 years of age. As noted above, there is wide variation in published estimates of the penetrance of CDKN2A mutations.
Pathology
The histopathologic characteristics of melanoma in FAMMM kindreds are not different from those seen in sporadic cases of melanoma and, thus, are not useful in diagnosing the syndrome. Superficial spreading melanoma (SSM) and nodular melanoma are the most frequently encountered histological melanoma subtypes in patients with CDKN2A mutations, which is consistent with the relative early age of onset.
Diagnosis
Definition
FAMMM has been described by multiple authors and institutions, and various definitions have been adopted.[5] According to Newton et al., a scoring system allotting one point per feature establishes FAMMM with scores greater than or equal to 3. The features include: 1) two or more clinically atypical nevi, 2) more than 100 nevi in patients between 20 and 50 years of age, 3) more than 50 nevi in patients under 20 years of age or more than 50 years of age, 4) more than one nevus in buttocks or instep, 5) nevi on the anterior scalp, 6) one or more pigmented lesions in the iris.
The Classical (1990) definition uses the following criteria: 1) 100 or more melanocytic nevi, 2) one or more melanocytic nevi greater than or equal to 8mm in its largest diameter, and 3) one or more clinically atypical melanocytic nevi.
The National Institutes of Health (NIH) Consensus 1992 definition, which is still controversial, requires a family history of melanoma, in addition to a large number of melanocytic nevi (often greater than 50) and melanocytic nevi that present certain histological features.
Management
Screening for melanoma in FAMMM kindreds should begin at age 10 with a baseline total body skin examination including scalp, eyes, oral mucosa, genital area, and nail, as family members may develop melanoma in their early teens. Monthly self-performed skin examinations and early referral to a dermatologist for monitoring are recommended interventions.
Treatment approaches such as removal of the largest dysplastic nevus or all of the dysplastic nevi have not been shown to appreciably reduce the risk of developing melanoma and are not cost-effective; therefore, these approaches are not recommended. Similarly, biopsy of multiple pigmented dysplastic nevi is not recommended and biopsy should be limited to specific nevi with appearance concerning for melanoma.
At Mayo Clinic, FAMMM patients with a confirmed mutation and family history of pancreatic cancer are offered screening with either high-resolution pancreatic protocol CT, MRI, or endoscopic ultrasound starting at age 50 or 10 years younger than the earliest family member with pancreas cancer. They are counseled on the lack of evidence-based data to support screening, and on the limitations of our current technology to detect a lesion at a stage amenable to therapy.[4]
History
In 1820 Norris reported for the first time a case of what is now recognized as FAMMM (12). He described a 59-year-old man with melanoma, a high total body mole count, and family history of the same.[6]
See also
- Dysplastic nevus
- List of genes mutated in pigmented cutaneous lesions
References
- "Melanoma". www.clevelandclinicmeded.com.
- James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: Clinical Dermatology. Saunders Elsevier. ISBN 978-0-7216-2921-6.
- Czajkowski, R., FAMMM Syndrome: Pathogenesis and Management, Dermatologic Surgery, Vol. 30 Issue 2 p2, p291-296, 2004.
- "FAMMM : In Familial Cancer Syndromes. DL Riegert-Johnson and others. NCBI 2009". Retrieved 2009-07-21.
- Silva JH, Sá BC, Avila AL, Landman G, Duprat Neto JP (2011). "Atypical mole syndrome and dysplastic nevi: identification of populations at risk for developing melanoma - review article". Clinics (Sao Paulo). 66 (3): 493–9. doi:10.1590/s1807-59322011000300023. PMC 3072014. PMID 21552679.
- "FAMMM : In Familial Cancer Syndromes. DL Riegert-Johnson and others. NCBI 2009". Retrieved 2009-07-21.
External links
| Classification |
|---|