Batten disease
Batten disease is a fatal disease of the nervous system that typically begins in childhood.[1] Onset of symptoms is usually between 5 and 10 years of age.[1] Often, it is autosomal recessive. It is the common name for a group of disorders called the neuronal ceroid lipofuscinoses (NCLs).[1]
| Batten disease | |
|---|---|
| Other names | Spielmeyer–Vogt–Sjögren–Batten disease, Batten–Mayou disease, Vogt–Spielmeyer disease |
| Specialty | Endocrinology |
| Usual onset | 5 to 10 years old[1] |
| Causes | Genetic[1] |
| Frequency | 2 to 4 per 100,000 births in the US[1] |
Although Batten disease is usually regarded as the juvenile form of NCL (or "type 3"), some physicians use the term Batten disease to describe all forms of NCL. Historically, the NCLs were classified by age of disease onset as infantile NCL (INCL), late infantile NCL (LINCL), juvenile NCL (JNCL) or adult NCL (ANCL).[2] At least 20 genes have been identified in association with Batten disease, but juvenile NCL, the most prevalent form of Batten disease, has been linked to mutations in the CLN3 gene.[3][4]It was first described in 1903.[1]
Signs and symptoms
Early signs and symptoms of the disorder usually appear around ages 2–10, with gradual onset of vision problems or seizures. Early signs may be subtle personality and behavioral changes, slow learning or regression, repetitive speech or echolalia, clumsiness or stumbling. Slowing head growth in the infantile form, poor circulation in lower extremities (legs and feet), decreased body fat and muscle mass, curvature of the spine, hyperventilation and/or breath-holding spells, teeth grinding and constipation may occur.
Over time, affected children suffer mental impairment, worsening seizures and progressive loss of sight, speech and motor skills. Batten disease is a terminal disease; life expectancy varies depending on the type or variation.
Females with juvenile Batten disease show first symptoms a year later than males, but on average die a year sooner.[5]
Cause
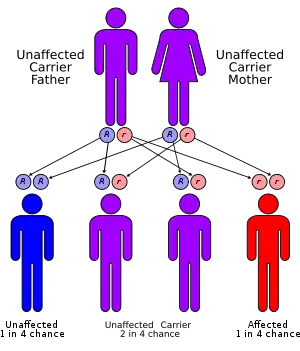
NCLs are a family of diseases that are inherited in an autosomal recessive manner. Collectively referred to as Batten disease, NCLs are responsible for most paediatric neurodegenerative diseases. The specific type of NCL is characterized by the age of symptomatic onset and genetic mutation involved. Currently, mutations in ten genes are believed to lead to the development of Batten disease; 'the incidence is as high as one in 12,500 live births'.[6]
NCL diseases
- Infantile neuronal ceroid (INCL): CLN1 encodes for the protein PPT1 which functions as a lysosomal enzyme.[7]
- Late infantile NCL (LINCL): CLN2 encodes for the protein TPP1 which serves as a lysosomal enzyme.[7] Life expectancy is between eight and twelve years of age. [8]
- Juvenile NCL (JNCL): CLN3 encodes for CLN3, a lysosomal transmembrane protein.[7]
- Adult NCL: CLN4 has no known associated protein.[7]
- Finnish variant of late infantile NCL (fLINCL): CLN5 encodes for CLN5, a soluble lysosomal protein.[7]
- Variant of the late infantile NCL: CLN6 encodes for the protein CLN6, which serves as a transmembrane protein of the endoplasmic reticulum.[7]
- Turkish variant of late infantile NCL: CLN7 or MFSD8, encodes for MFSD8, which functions as a lysosomal transmembrane protein.[7]
- Northern epilepsy: CLN8 encodes for CLN8, a transmembrane protein of the endoplasmic reticulum.[7]
- Late infantile NCL: CLN10 or CTSD encodes for CTSD, which is a lysosomal protein which a variety of functions.[7]
- Infantile osteopetrosis: CLCN7 encodes for CLCN7.[7]
Juvenile NCL: CLN3 mutation
The CLN3 gene is located on the short arm of chromosome 16 at gene position 12.1 (16p12.1), and mutations within this gene are the major cause of juvenile NCL. More specifically, 73% of Batten disease cases are due to a 1.02-kb deletion within this gene, CLN3, which causes a frameshift which produces a truncated mutant gene product of only 181 amino acids in length when compared to the wild-type gene product of 438 amino acids in length. Normal-functioning CLN3 encodes for a hydrophobic transmembrane protein that is mainly localized to the lysosome; however, the 181 amino acid mutant gene product was instead found to primarily localize to the endoplasmic reticulum and Golgi apparatus. The precise function of the CLN3 gene product remains unknown.[6]
Diagnosis
Batten disease is rare; misdiagnosis may lead to increased medical expenses, family stress, and the chance of using incorrect forms of treatment, which may exacerbate the patient's condition. Nevertheless, Batten disease can be diagnosed if properly detected. Vision impairment is the most common observable symptom of the disease. Occurrences in children are more prevalent than occurrences in adolescents or adults. Children or adults suspected of having Batten disease should initially see an optometrist or ophthalmologist. A fundus eye examination that aids in the detection of common vision impairment abnormalities, such as granularity of the retinal pigment epithelium in the central macula will be performed. Though it is also seen in a variety of other diseases, a loss of ocular cells is a warning sign of Batten disease. If Batten disease is the suspected diagnosis, a variety of tests is conducted to help accurately confirm the diagnosis, including:
- Blood or urine tests can help detect abnormalities that may indicate Batten disease. For example, elevated levels of dolichol in urine have been found in many individuals with NCL. The presence of vacuolated lymphocytes—white blood cells that contain holes or cavities (observed by microscopic analysis of blood smears)—when combined with other findings that indicate NCL, is suggestive for the juvenile form caused by CLN3 mutations.[9]
- Skin or tissue sampling is performed by extracting a small piece of tissue, which then is examined under an electron microscope. This can allow physicians to detect typical NCL deposits. These deposits are common in tissues such as skin, muscle, conjunctiva, and rectum.[9] This diagnostic technique is useful, but other invasive tests are more reliable for diagnosing Batten disease.
- Electroencephalogram (EEG) is a technique that uses special probes attached on to the individual's scalp. It records electrical currents/signals, which allow medical experts to analyze electrical pattern activity in the brain. EEG assists in observing if the patient has seizures.[9]
- Electrical studies of the eyes are used, because as mentioned, vision loss is the most common characteristic of Batten disease. Visual-evoked responses and electroretinograms are effective tests for detecting various eye conditions common in childhood NCLs.
- Computed tomography (CT) or magnetic resonance imaging (MRI) are diagnostic imaging tests that allow physicians to better visualize the appearance of the brain. MRI imaging test uses magnetic fields and radio waves to help create images of the brain. CT scan uses x-rays and computers to create a detailed image of the brain's tissues and structures. Both diagnostic imaging tests can help reveal brain areas that are decaying, or atrophic, in persons with NCL.[9]
- Measurement of enzyme activity specific to Batten disease may help confirm certain diagnoses caused by different mutations. Elevated levels of palmitoyl-protein thioesterase is involved in CLN1. Acid protease is involved in CLN2. Cathepsin D is involved in CLN10.[9]
- DNA analysis can be used to help confirm the diagnosis of Batten disease. When the mutation is known, DNA analysis can also be used to detect unaffected carriers of this condition for genetic counseling. If a family mutation has not previously been identified or if the common mutations are not present, recent molecular advances have made it possible to sequence all of the known NCL genes, increasing the chances of finding the responsible mutation(s).[9]
Treatment
Batten disease is a terminal illness; the FDA has approved Brineura (cerliponase alfa) as a treatment for a specific form of Batten disease. Brineura is the first FDA-approved treatment to slow loss of walking ability (ambulation) in symptomatic pediatric patients 3 years of age and older with late infantile neuronal ceroid lipofuscinosis type 2 (CLN2), also known as tripeptidyl peptidase-1 (TPP1) deficiency. Palliative treatment is symptomatic and supportive. One drug, an antisense oligonucleotide, milasen,[10] described in The New England Journal of Medicine,[11] is believed to be the first “custom” treatment for a genetic disease. It is named after Mila Makovec, the only patient who may ever take it. (It could help another exceedingly rare patient who has the same mutation as Mila.)
History
Batten disease is named after the British pediatrician Frederick Batten, who first described it in 1903.[12][13] Also known as Spielmeyer-Vogt-Sjögren-Batten disease, it is the most common form of a group of disorders called neuronal ceroid lipofuscinosis (NCL). Although Batten disease is usually regarded as the juvenile form of NCL, some physicians use the term Batten disease to describe all forms of NCL.
Research

In June 1987, a phase-I clinical trial was launched at Weill Cornell Medical College of Cornell University to study a gene therapy method for treatment of the signs and symptoms of LINCL. The experimental drug works by delivering a gene transfer vector called AAV2CUhCLN2 to the brain.[14] Although the trial is not matched, randomized, or blinded and lacked a contemporaneous placebo/sham control group, assessment of the primary outcome variable suggests a slowing of progression of LINCL in the treated children.[15]
Researchers believe the neurological deficits common in JNCL could be due to overactive AMPA receptors in the cerebellum. To test this hypothesis, researchers administered AMPA antagonist drugs into affected mice. The motor skills of the affected mice showed significant improvement after the antagonist treatment, which supported the hypothesis that the neurological deficits in JNCL are due to overactive AMPA receptors. This research could eventually help to alleviate neurological deficits of JNCL in humans.[16]
In November 2006, after receiving FDA clearance, neurosurgeon Nathan Selden, pediatrician Bob Steiner, and colleagues at Doernbecher Children's Hospital at Oregon Health and Science University began a clinical study in which purified neural stem cells were injected into the brain of Daniel Kerner, a six-year-old child with Batten disease, who had lost the ability to walk and talk. This patient was the first of six to receive the injection of a stem cell product from StemCells Inc., a Palo Alto biotech company. These are believed to be the first-ever transplants of fetal stem cells into the human brain.[17] By early December, the child had recovered well enough to return home, and some signs of speech returning were reported.[18] [19] The main goal of phase-I clinical trials, however, was to investigate the safety of transplantation. Overall, the phase-I data demonstrated that high doses of human neural stem cells, delivered by a direct transplantation procedure into multiple sites within the brain, followed by 12 months of immunosuppression, were well tolerated by all six patients enrolled in the trial. The patients’ medical, neurological, and neuropsychological conditions, following transplantation, appeared consistent with the normal course of the disease. Daniel Kerner died on August 20th, 2009.
In 2010, Cherie and Jim Flores donated $2 million, the biggest gift in Batten disease research history, and the Beyond Batten Disease Foundation contributed $500,000 to establish laboratories for Italian researchers Drs. Ballabio, Sardiello and their colleagues at the Jan and Dan Duncan Neurological Research Institute of Texas Children’s Hospital.[14]
During 2011, the first controlled clinical trials began with the University of Rochester for a treatment for Batten Disease.[15] The trial included 30 patients who were experiencing signs of the disease in the hope of slowing its progress.
In November 2013, Weill Medical College of Cornell University began recruiting participants for a safety study of a gene transfer vector,[16] described as a non-randomised safety and efficacy trial. As part of a trial began by University of Rochester in March 2014. Mycophenolate mofetil is being tested to determine its efficacy and safety using a gene transfer vector.
In complex diseases such as Batten, therapies that address multiple aspects of the disease at the same time have the potential for higher impact than those focusing on one aspect.“The use of several treatment strategies might offer additional benefits to patients with neurodegenerative disease, but the benefits of this approach must be weighed carefully against the additional adverse effects that combined treatments might bring,” the researchers wrote. The medical team also noted that “over the past two decades, scientists and clinicians within the Batten disease community have worked to ensure that tools are in place to enable progress towards effective treatments at an unprecedented pace. ”Recent progress in Batten disease research offers hope that efficient and targeted therapies will be available soon, the researchers said, noting that the “Batten disease research community is becoming a model of how effective, efficient rare disease research can be accomplished by working together.”
One drug, an antisense oligonucleotide, milasen[10], described in The New England Journal of Medicine[11], is believed to be the first “custom” treatment for a genetic disease. It is named after the patient for who it was designed and the only person who may ever take it, Mila Makovec, who has Batten CLN7. More about this story can be found on the Mila's Miracle Foundation website.
See also
References
- "Batten Disease Fact Sheet | National Institute of Neurological Disorders and Stroke". www.ninds.nih.gov. Retrieved 30 November 2020.
- Hobert JA, Dawson G (October 2006). "Neuronal ceroid lipofuscinoses therapeutic strategies: past, present and future". Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1762 (10): 945–53. doi:10.1016/j.bbadis.2006.08.004. PMID 17049436.
- Rakheja D, Narayan SB, Bennett MJ (September 2007). "Juvenile neuronal ceroid-lipofuscinosis (Batten disease): a brief review and update". Current Molecular Medicine. 7 (6): 603–8. doi:10.2174/156652407781695729. PMID 17896996.
- Cooper JD (June 2008). "Moving towards therapies for juvenile Batten disease?". Experimental Neurology. 211 (2): 329–31. doi:10.1016/j.expneurol.2008.02.016. PMID 18400221. S2CID 32126291.
- Cialone J, Adams H, Augustine EF, et al. (May 2012). "Females experience a more severe disease course in Batten disease". Journal of Inherited Metabolic Disease. 35 (3): 549–55. doi:10.1007/s10545-011-9421-6. PMC 3320704. PMID 22167274.
- Jill M. Weimer; Elizabeth Kriscenski-Perry; Yasser Elshatory; David A. Pearce (2002). "The Neuronal Ceroid Lipofuscinoses: Mutations in Different Proteins Result in Similar Disease". NeuroMolecular Medicine. 1 (2): 111–124. doi:10.1385/nmm:1:2:111. PMID 12025857. S2CID 33921126.
- Jalanko, Anu; Braulke, Thomas (2009). "Neuronal ceroid lipofuscinoses". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1793 (4): 697–709. doi:10.1016/j.bbamcr.2008.11.004. PMID 19084560.
- "Warren Recognizes Batten Disease Awareness Day". June 4, 2018. Retrieved May 30, 2020.
- "Noah's Hope - Causes and Symptoms of Batten Disease". www.noahshope.com. Archived from the original on 2016-11-23. Retrieved 2016-11-22.
- Chen, Angus (October 15, 2019). "A Drug Was Made For Just One Child, Raising Hopes About Future Of Tailored Medicine". Retrieved May 30, 2020.
- Kim, Jinkuk; Hu, Chunguang; Moufawad El Achkar, Christelle; Black, Lauren E.; Douville, Julie; Larson, Austin; Pendergast, Mary K.; Goldkind, Sara F.; Lee, Eunjung A.; Kuniholm, Ashley; Soucy, Aubrie; Vaze, Jai; Belur, Nandkishore R.; Fredriksen, Kristina; Stojkovska, Iva; Tsytsykova, Alla; Armant, Myriam; Didonato, Renata L.; Choi, Jaejoon; Cornelissen, Laura; Pereira, Luis M.; Augustine, Erika F.; Genetti, Casie A.; Dies, Kira; Barton, Brenda; Williams, Lucinda; Goodlett, Benjamin D.; Riley, Bobbie L.; Pasternak, Amy; et al. (2019). "Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease". New England Journal of Medicine. 381 (17): 1644–1652. doi:10.1056/NEJMoa1813279. PMC 6961983. PMID 31597037.
- synd/7 at Who Named It?
- Batten FE (1902). "Cerebral degeneration with symmetrical changes in the maculae in two members of a family". Transactions of the Ophthalmological Societies of the United Kingdom. 23: 386–90.
- Clinical trial number NCT00151216 for "Safety Study of a Gene Transfer Vector for Children With Late Infantile Neuronal Ceroid Lipofuscinosis" at ClinicalTrials.gov
- Worgall S, Sondhi D, Hackett NR, et al. (May 2008). "Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA". Human Gene Therapy. 19 (5): 463–74. CiteSeerX 10.1.1.553.872. doi:10.1089/hum.2008.022. PMID 18473686.
- Kovács, Attila D.; Pearce, David A. (2008-01-01). "Attenuation of AMPA receptor activity improves motor skills in a mouse model of juvenile Batten disease". Experimental Neurology. The Role of α-synuclein in the Pathogenesis of Parkinson's Disease / Gene Therapy for Parkinson's. 209 (1): 288–291. doi:10.1016/j.expneurol.2007.09.012. PMC 4418195. PMID 17963751.
- "A stem cell first at OHSU Archived 2012-02-06 at the Wayback Machine" The Portland Tribune, Nov 24, 2006
- http://www.technologyreview.com/read_article.aspx?id=17888&ch=biotech%5B%5D%5B%5D
- "Archived copy". Archived from the original on 2015-10-24. Retrieved 2015-09-21.CS1 maint: archived copy as title (link)
External links
| Wikimedia Commons has media related to Batten disease. |
- Batten disease at NINDS
- GeneReviews/NCBI/NIH/UW entry on Neuronal Ceroid-Lipofuscinosis
- Batten FE, Mayou MS (1915). "Family Cerebral Degeneration with Macular Changes". Proceedings of the Royal Society of Medicine. 8 (Sect Ophthalmol): 70–90. doi:10.1177/003591571500801624. PMC 2003604. PMID 19978990.
| Classification | |
|---|---|
| External resources |