Triple-stranded DNA
Triple-stranded DNA (also known as H-DNA or Triplex-DNA) is a DNA structure in which three oligonucleotides wind around each other and form a triple helix. In triple-stranded DNA, the third strand binds to a B-form DNA (via Watson–Crick base-pairing) double helix by forming Hoogsteen base pairs or reversed Hoogsteen hydrogen bonds.
.png.webp)
Structure

Hoogsteen base pairing
A thymine (T) nucleobase can bind to a Watson–Crick base-pairing of T-A by forming a Hoogsteen hydrogen bond. The thymine hydrogen bonds with the adenosine (A) of the original double-stranded DNA to create a T-A*T base-triplet.[1] Under acidic conditions, a protonated cytosine, represented as C+, can form a base-triplet with a C-G pair through Hoogsteen base-pairing, forming C-G*C+. The TA*T and CG*C+ base pairs are the most stabilized triplet-base pairs that can form, while a TA*G and CG*G are the most destabilized triplet-base pairs.[2]
Intermolecular and intramolecular interactions
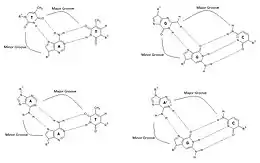
There are two classes of triplex DNA: intermolecular and intramolecular formations. An intermolecular triplex refers to triplex formation between a duplex and a different (third) strand of DNA. The third strand can either be from a neighboring chromosome or a triplex forming oligonucleotide (TFO). Intramolecular triplex DNA is formed from a duplex with homopurine and homopyrimidine strands with mirror repeat symmetry.[3] The degree of supercoiling in DNA influences the amount of intramolecular triplex formation that occurs.[4] There are two different types of intramolecular triplex DNA: H-DNA and H*-DNA. Formation of H-DNA is stabilized under acidic conditions and in the presence of divalent cations such as Mg2+. In this conformation, the homopyrimidine strand in the duplex bends back to bind to the purine strand in a parallel fashion. The base triads used to stabilize this conformation are T-A*T and C-G*A+. The cytosine of this base triad needs to be protonated in order to form this intramolecular triple helix, which is why this conformation is stabilized under acidic conditions.[5] H*-DNA has favorable formation conditions at neutral pH and in the presence of divalent cations.[4] This intramolecular conformation is formed from the binding of the homopurine and purine strand of the duplex in an antiparallel fashion. It is stabilized by T-A*A and C-G*G base triplets.[3][5]
Function
Triplex forming oligonucleotides (TFO)
TFOs are short (≈15-25 nt) nucleic acid strands that bind in the major groove of double-stranded DNA to form intramolecular triplex DNA structures. There is some evidence that they are also able to modulate gene activity in vivo. In peptide nucleic acid (PNA), the sugar-phosphate backbone of DNA is replaced with a protein-like backbone. PNAs form P-loops while interacting with duplex DNA, forming a triplex with one strand of DNA while displacing the other. Very unusual recombination or parallel triplexes, or R-DNA, have been assumed to form under RecA protein in the course of homologous recombination.[6]
TFOs bind specifically to homopurine-homopyrimidine regions that are often common in promoter and intron sequences of genes, influencing cell signaling.[7] TFOs can inhibit transcription by binding with high specificity to the DNA helix, thereby blocking the binding and function of transcription factors for particular sequences. By introducing TFOs into a cell (through transfection or other means), the expression of certain genes can be controlled.[8] This application has novel implications in site-specific mutagenesis and gene therapy. In human prostate cancer cells, a transcription factor Ets2 is over-expressed and thought to drive forward the growth and survival of cells in such excess. Carbone et al. designed a sequence-specific TFO to the Ets2 promoter sequence that down-regulated the gene expression and led to a slowing of cell growth and cell death.[9] Changxian et al. have also presented a TFO targeting the promoter sequence of bcl-2, a gene inhibiting apoptosis.[10]
The observed inhibition of transcription can also have negative health effects like its role in the recessive, autosomal gene for Friedreich’s Ataxia.[11] In Fredrick’s Ataxia, triplex DNA formation impairs the expression of intron 1 of the FXN gene. This results in the degeneration of the nervous system and spinal cord, impairing the movement of the limbs.[12] To combat this triplex instability, nucleotide excision repair proteins (NERs) have been shown to recognize and repair triple-stranded DNA structures, reinstating full availability of the previously inhibited and unstable gene.[13]
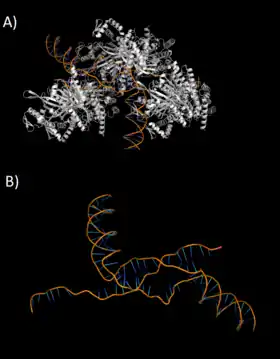
Peptide nucleic acids (PNA)
Peptide nucleic acids are synthetic oligonucleotides that resist protease degradation and are used to induce repair at site specific triplex formation regions on DNA genomic sites. PNAs are able to bind with high affinity and sequence specificity to a complementary DNA sequence through Watson-Crick base pairing binding and are able to form triple helices through parallel orientation Hoogsteen bonds with the PNA facing the 5’-end of the DNA strand.[14] The PNA-DNA triplex are stable because PNAs consist of a neutrally charged pseudopeptide backbone which binds to bind to the double stranded DNA (dsDNA) sequence.[15] Similar to homopyrimidine in TFOs, homopyrimidine in PNAs are able to form a bond with the complementary homopurine in target sequence of the dsDNA. These DNA analogues are able to bind to dsDNA by exploiting ambient DNA conditions and different predicting modes of recognition. This is different from TFOs which bind though the major groove recognition of the dsDNA.[14]
One of the predicting modes of recognition used for recognition is through a duplex invasion.[16][15] Within mixed A–T/G–C dsDNA sequence is targeted by a pair of pseudo-complementary (pc) PNAs which are able to bind to dsDNAs via double invasion through the simultaneous formation of diaminopurine (D) and thiouracil (Us) which substitute for adenine and thymine, respectively.[16] The pc PNA pair form a D-T and Us -A and G-C or C-G Watson-Crick paired PNA-DNA helix with each of complementary DNA strands. Another form of recognized duplex invasion at targeted sequence can occur in dsDNA containing mixed T–C sequences.[17] This form of duplex invasion is achieved through a complimentary sequence of homopurine PNA oligomers. This triplex is formed from a PNA-DNA hybrid that binds anti-parallel with the complementary DNA sequence and results in a displaced non-complimentary DNA strand.[15]
Additionally, PNA can be modified to form “clamp” triplex structures at the target site.[16] One type of “clamp” formed is a bis-PNA structure, in which two PNA molecules are held together by a flexible linker such as 8-amino-3,6-dioxaoctanoic acid (O).[18] The bis-PNA structure forms a PNA-DNA-PNA triplex at the target site, where one strand forms Watson-Crick base pairs with DNA in an antiparallel orientation and the other strand forms Hoogsteen base pairs with the homopurine DNA strand in the DNA-PNA duplex.[17] A tail clamp PNA (tcPNA) is also another form of triplex clamp that can also be formed. TcPNAs contain an extended 5-10 bp tail that forms a PNA/DNA duplex in addition to a PNA-DNA-PNA “clamp”. This allows for more specified PNA binding without the need for a homopyrimidie/pyridine stretch.[15] These clamp structures had been shown to have high affinity and specificity. The addition of lysine residues to either or both ends of PNA’s could be used to increase cellular uptake and binding.[16]
Genetic regulation
Triple-stranded DNA has been implicated in the regulation of several genes. For instance, the c-myc gene has been extensively mutated to examine the role that triplex DNA, versus the linear sequence, plays in gene regulation. A c-myc promoter element, termed the nuclease-sensitive element or NSE, can form tandem intramolecular triplexes of the H-DNA type and has a repetitive sequence motif (ACCCTCCCC)4. The mutated NSE was examined for transcriptional activity and for its intra- and intermolecular triplex-forming ability. The transcriptional activity of mutant NSEs can be predicted by the element's ability to form H-DNA and not by repeat number, position, or the number of mutant base pairs. DNA may therefore be a dynamic participant in the transcription of the c-myc gene.[19]
Gene expression
According to several published articles, H-DNA has the ability to regulate gene expression depending on factors such as location and sequences in proximity. Although intergenic regions of the prokaryotic genome have shown low traces of naturally occurring H-DNA or triplex motifs, H-DNA structures have shown to be more prevalent in the eukaryotic genome. H-DNA has been show to be especially abundant in mammalian cells including humans (1 in every 50,000 bp).[14] Genetic sequences involved in gene regulation are typically found in the promoter regions of the eukaryotic genome.[14]
Consequently, the promoter region has displayed the ability to form H-DNA with a higher frequency.[14] A bioinformatic analysis of the S. cerevisiae genome observed the occurrence of H-DNA and other triplate DNA motifs in four organizational regions: introns, exons, promoter regions and miscellaneous regions. The bioinformatic displayed a total of 148 H-DNA or triplet DNA possible structures. The promoter region accounted for the higher frequency with 71 triplate structures, while the exons accounted for 57 triplate structures and the introns and miscellaneous accounted for 2 and 18 structures.[20]
In vitro and in vivo studies of eukaryotic genome expression resulted in one of three results: up regulation, down regulation, or no change in the presence of H-DNA motifs.[14] Kato et. al. reported upregulation expression of lacZ, when H-DNA was introduced to the B-lactamase promoter.[21][14] On the other hand, a similar study (Brachmachari et al.) reported no statistically significant inhibition of the lacZ reporter gene when H-DNA was inserted into the genome of mammalian COS cells.[14] Although studies suggest regulation of H-DNA, the mechanism is still under investigation. Potaman et al. associates the mechanism of gene regulation to the interactions between the H-DNA and the TATA box found in the promoter region of Na,K-ATPase. In H-DNA formations adjacent to a TATA box, the H-DNA structure destabilizes the T-A bonds essential for transcription. The interference with the TATA box inhibits the transcriptional machinery and transcription initiation which interferes with gene expression.[14][22] Other mechanisms associated with the genomic expression of a genetic sequence in the presence of H-DNA involves TFOs. In vitro studies have highlighted a decrease in gene expression in the presence of TFOs in mammalian cells.[23] Another possible mechanism presented by Valentina et al. suggest the 13-mer AG motif oligonucleotide triplex complex (TFO complex) downregulates the transcription of mRNA through competitive inhibition.[24] Direct inhibition of gene expression from H-DNA is key to mutagenesis, replication inhibition, and even DNA recombination in the genome.[14]
Recombination
H-DNA motifs have been shown to stimulate homologous recombination with different mechanisms. Initial implications for the role of H-DNA in recombination came in the early 1990s when observing RecA, a bacterial DNA recombination protein composed of triple-helix DNA. RecA exhibits enzymatic activity essential for recombination.[6][25] Homologous recombination involving H-DNA motifs have also been found in eukaryotes. RadA, a homologous protein to RecA, has been shown to have the same enzymatic activity in recombination as RecA.[26] The protein has the ability to promote and exchange homologous strands through parallel triple stranded helices.[27][28] The single stranded DNA (ssDNA) and complementary double stranded DNA (dsDNA) will form a D-loop structure.[29][14] Another possible mechanism for RecA involves the ssDNA from two separate H-DNA structures to form Watson-Crick base pairs. The new structure is known as a Holliday junction, an intermediate in homologous recombination.[14] H-DNA is also found in other forms of recombination. In mammalian cells, H-DNA-sequences displayed a high frequency of recombination. For example, a study conducted on myeloma cell line of mice found H-DNA structures in Cγ2a and Cγ2b, which participate in sister chromatid exchange.[14]
Biological Implications
Genetic Instability
Considerable research has been funneled into the biological implications relating to the presence of H-DNA in the major breakpoint regions (Mbr) and double-strand-breakpoints of certain genes. Recent work has linked the presence of non-B-DNA structures with cases of genetic instability.[30]
Polypurine mirror-repeat H-DNA forming sequences were found neighboring the P1 promoter of the c-MYC gene and are associated with the major breakpoint hotspots of this region. Cases of genetic instability were also observed in the F1 offspring of transgenic mice after incorporation of human H-DNA-forming sequences paired with Z-DNA sequences into their genomes where no instability was previously reported.[31] Additionally, formation of R.R.Y. H-DNA conformations have been observed at the Mbr of the bcl-2 gene. Formation of these structures has been posited to cause the t(14;18) translocation observed in many cancers and most follicular lymphomas. This observation has led to research that indicated a substantial decrease in translocation events can be observed after blocking the formation of H-DNA by altering the sequence of this region slightly.[31][32] Long tracts of GAA·TTC have also been observed to form very stable H-DNA structures. Interactions between these two H-DNA structures, termed sticky DNA, has been shown to interrupt transcription of the X25, or frataxin gene. As decreased levels of the protein frataxin is associated with Friedreich's ataxia, formation of this instability has been suggested to be the basis for this genetic disease.[33][34]
Additionally, H-DNA has been shown to cause mutations related to critical cellular processes like DNA replication and transcription.[35] The importance of these processes for survival has led to the development of complex DNA repair mechanisms that allow cells to recognize and fix DNA damage. Non-canonical DNA structures can be perceived as damage by the cell, and recent work has shown an increased prevalence of mutations near non-B-DNA-forming sequences.[35] Some of these mutations are due to the interactions between H-DNA and the enzymes involved in DNA replication and transcription, where H-DNA interferes with these processes and triggers various DNA repair mechanisms. This can cause genetic instability and implicates H-DNA in cancer formation.[35]
DNA Replication
DNA replication has been shown to affect the function of various DNA repair enzymes. H-DNA formation involves the formation of single-stranded DNA (ssDNA), which is more susceptible to attack by nucleases.[35] Various nucleases have been shown to interact with H-DNA in a replication-dependent or replication-independent manner.[35]
A study using human cells found that the nucleotide excision repair (NER) nucleases ERCC1-XPF and ERCC1-XPG induced genetic instability.[36] These enzymes cleave H-DNA at the loop formed by the two Hoogsteen hydrogen-bonded strands and the 5' end of the other Watson-Crick hydrogen-bonded strand, respectively.[36] This cleavage has been shown to induce large deletions that cause double strand breaks (DSBs) in DNA that can lead to genetic instability.[35][36] In cells deficient in ERCC1-XPF and ERCC1-XPG, these deletions were less prevalent near H-DNA forming sequences.[36] Additionally, more mutations were found in ERCC1-XPF and ERCC1-XPG deficient cells in the absence of DNA replication, which suggests they process H-DNA in a replication-independent manner.[36]
Alternatively, the DNA-replication repair nuclease FEN1 was found to suppress genetic instability.[36] Similar to ERCC1-XPG, FEN1 cleaves H-DNA at the 5' end of the strand not involved in Hoogsteen hydrogen-bonding.[36] HeLa cells deficient in FEN1 showed higher prevalence of deletions near H-DNA forming sequences, but H-DNA induced mutagenesis was more pronounced in FEN1 deficient cells in the presence of DNA replication.[36] This suggests FEN1 suppresses H-DNA-induced mutagenesis in a replication-dependent manner.[36]
H-DNA has been implicated in human cancer etiology because of the prevalence of H-DNA-forming sequences near translocation breakpoints in cancer genomes.[36] Replication-mediated nuclease activity with H-DNA highlights another way H-DNA-induced mutagenesis and lead to cancer growth.
Transcription
H-DNA forming sequences can also cause genetic instability by interfering with and stopping transcription prematurely.[35] The DNA unwinding involved in transcription makes it more susceptible to damage. In transcription-coupled repair (TCR), a lesion on the template strand of DNA stops the function of RNA polymerase and signals TCR factors to resolve the damage by excising it.[37] H-DNA can be perceived as one of these lesions.
A study observing transcription by T7 RNA polymerase on a stable H-DNA-forming sequence analog found transcription blockage at the duplex-to-triplex junction. Here, the template strand was the central strand of the H-DNA, and the difficulty of disrupting its Watson-Crick and Hoogsteen hydrogen bonds stopped transcription from progressing.[38]
When transcription by T7 was observed on the P0 promoter of the c-MYC gene, the shortened transcription products that were found indicated that transcription was stopped in close proximity to the H-DNA forming sequence downstream of the promoter. Formation of H-DNA in this region prevents T7 from traveling down the template strand because of the steric hindrance it causes. This stops transcription and signals for TCR factors to come resolve the H-DNA, which results in DNA excision that can cause genetic instability.[37] The mirror symmetry and prevalence of guanine residues in the c-MYC gene gives it a high propensity for non-canonical DNA structure formation.[39] This coupled with the activity of TCR factors during transcription makes it highly mutagenic, with it playing a role in the development of Burkitt lymphoma and leukemia.[37][39]
Applications
The triple-stranded DNA regions can be generated through the association of Triplex Forming Oligonucleotides (TFO) and Peptide Nucleic Acids (PNAs). Historically, TFO binding has been shown to inhibit transcription, replication, and protein binding to DNA.[16] TFOs tethered to mutagens have also been shown to promote DNA damage and induce mutagenesis.[14] Although TFO have been known to hinder transcription and replication of DNA, recent studies have shown that TFO can be utilized to mediate site specific gene modifications both in vitro and in vivo.[16] Another recent study has also shown that TFOs can be used for suppression of oncogenes and proto-oncogenes to reduce cancer cell growth. For example, a recent study has used TFOs to reduce cellular death in hepatoma cells through the decreasing the expression of MET.
PNA TFOs have the ability to enhance recombination frequencies, leading to targeted, specific editing of genes. The PNA-DNA-PNA triplex helix is able to be recognized by the cell’s own DNA repair mechanism, which sensitizes the surrounding DNA for homologous recombination. In order for a site-specific PNA structure to mediate recombination within a DNA sequence, a bis-PNA structure can be coupled with a 40nt DNA fragment that is homologous to an adjacent region on the target gene.[17] The linking of a TFO to a donor DNA strand has been shown to induce recombination of the targeted gene and the adjacent gene target region.[40] The mechanism for this form of recombination and repair have been linked to the nucleotide excision repair (NER) pathway playing a role in recognizing and repairing triplex structures.[17][16] Multiple investigations suggests that the xeroderma pigmentosum group A (XPA) and replication protein A (RPA), which are NER factors, are able to bind specifically as a complex to cross-linked triplex structures. It is known that this mechanism alongside others play a role in recognizing and repairing triplex structures.
The in vivo delivery of TFOs has been a major barrier in using TFOs for gene modification.[41] One study on in vivo targeting of hematopoietic stem cells proposed a novel technique of conjugating PNA molecules with cell penetrating peptide (CPPs) alongside poly(lactic-co-glycolic acid) (PLGA) nanoparticles to enable 6 bp modifications in the CCR5 gene.[40] The editing of the CCR5 gene has been linked to HIV-1 resistance.[42] CPPs are proteins that are able to carry “cargo” such as small proteins or molecules successfully into cells. The PGLAs are biodegradable material that encapsulate PNA molecules as nanoparticles for site specific genome modifications.[40] The study found that the PNA-DNA PGLA nanoparticles were able to effectively edit the hematopoietic stem cells with lower toxicity and virus-free and the conjugation with CPP offered direct targeting of the genes for site-specific mutagenesis in the stem cells.
In a novel study of cystic fibrosis (CF) gene therapy, three tail-clamp peptide nucleic acids (PNAs) alongside donor DNA molecule were engineered to be delivered by nanoparticles to correct F508 del mutations on the cystic fibrosis transmembrane conductance regulator (CFTR) in human bronchial epithelial cells in vivo and in vitro.[43] The F508 del mutation is the most commonly occurring mutation which leads a person to have CF.[44] The F508 mutation leads to a loss of function of the CFTR, which is a plasma membrane chloride channel that is regulated by a cyclic-adenosine monophosphate(cAMP). In this study, they were able to create the novel treatment approach for CF through the use of nanoparticles to correct the F508 del CFTR mutation both in vitro in human bronchial epithelial (HBE) cells and in vivo in a CF mouse model which resulted in the appearance of CFTR-dependent chloride transport.[43]
History

Triple-stranded DNA structures were common hypotheses in the 1950s when scientists were struggling to discover DNA's true structural form. Watson and Crick (who later won the Nobel Prize for their double-helix model) originally considered a triple-helix model, as did Pauling and Corey, who published a proposal for their triple-helix model in 1953,[45][46] as well as fellow scientist Fraser.[47] However, Watson and Crick soon identified several problems with these models:
- Negatively charged phosphates near the axis repel each other, leaving the question of how the three-chain structure stays together.
- In a triple-helix model (specifically Pauling and Corey's model), some of the van der Waals distances appear to be too small.
Fraser's model differed from Pauling and Corey's in that in his model the phosphates are on the outside and the bases are on the inside, linked together by hydrogen bonds. However, Watson and Crick found Fraser's model to be too ill-defined to comment specifically on its inadequacies.
An alternative triple-stranded DNA structure was described in 1957.[48] Felsenfeld, Davies, and Rich predicted that if one strand contained only purines and the other strand only purines, the strand would undergo a conformational change to form a triple stranded DNA helix. The triple-stranded DNA (H-DNA) was predicted to be composed of one polypurine and two polypyrimidine strands.[6][48] It was thought to occur in only one in vivo biological process: as an intermediate product during the action of the E. coli recombination enzyme RecA.[48] Early models in the 1960s predicted the formation of complexes between polycetiylic and guanine oligonucleotides. The models suggested interactions known as Hoogsten pairing (non-Watson-Crick interactions) located in the major groove.[6] Shortly after, triple helices composed of one pyrimidine and two purine strands were predicted.[6] The discovery of in H-DNA stretches in supercoiled plasmids peaked modern interest in the potential function of triplex structures in living cells.[49] Additionally, it was soon found that homopyrimidine and some purine-rich oligonucleotide are able form a stable H-DNA structure with the homopurine-homopyrimidine binding sequence-specific structures on the DNA duplexes.[50]
References
- Rhee S, Han Z, Liu K, Miles HT, Davies DR (December 1999). "Structure of a triple helical DNA with a triplex-duplex junction". Biochemistry. 38 (51): 16810–5. doi:10.1021/bi991811m. PMID 10606513.
- Mergny JL, Sun JS, Rougée M, Montenay-Garestier T, Barcelo F, Chomilier J, Hélène C (October 1991). "Sequence specificity in triple-helix formation: experimental and theoretical studies of the effect of mismatches on triplex stability". Biochemistry. 30 (40): 9791–8. doi:10.1021/bi00104a031. PMID 1911764.
- Ussery DW, Sinden RR (June 1993). "Environmental influences on the in vivo level of intramolecular triplex DNA in Escherichia coli". Biochemistry. 32 (24): 6206–13. doi:10.1021/bi00075a013. PMID 8512930.
- Dayn A, Samadashwily GM, Mirkin SM (December 1992). "Intramolecular DNA triplexes: unusual sequence requirements and influence on DNA polymerization". Proceedings of the National Academy of Sciences of the United States of America. 89 (23): 11406–10. Bibcode:1992PNAS...8911406D. doi:10.1073/pnas.89.23.11406. PMC 50559. PMID 1454828.
- Lyamichev VI, Mirkin SM, Frank-Kamenetskii MD (February 1986). "Structures of homopurine-homopyrimidine tract in superhelical DNA". Journal of Biomolecular Structure & Dynamics. 3 (4): 667–9. doi:10.1080/07391102.1986.10508454. PMID 3271043.
- Frank-Kamenetskii MD, Mirkin SM (1995-01-01). "Triplex DNA structures". Annual Review of Biochemistry. 64: 65–95. doi:10.1146/annurev.bi.64.070195.000433. PMID 7574496. S2CID 21426188.
- Brázdová M, Tichý V, Helma R, Bažantová P, Polášková A, Krejčí A, et al. (2016). "p53 Specifically Binds Triplex DNA In Vitro and in Cells". PLOS ONE. 11 (12): e0167439. Bibcode:2016PLoSO..1167439B. doi:10.1371/journal.pone.0167439. PMC 5131957. PMID 27907175.
- Graham MK, Brown TR, Miller PS (April 2015). "Targeting the human androgen receptor gene with platinated triplex-forming oligonucleotides". Biochemistry. 54 (13): 2270–82. doi:10.1021/bi501565n. PMID 25768916.
- Carbone GM, Napoli S, Valentini A, Cavalli F, Watson DK, Catapano CV (2004-08-03). "Triplex DNA-mediated downregulation of Ets2 expression results in growth inhibition and apoptosis in human prostate cancer cells". Nucleic Acids Research. 32 (14): 4358–67. doi:10.1093/nar/gkh744. PMC 514370. PMID 15314206.
- Shen C, Rattat D, Buck A, Mehrke G, Polat B, Ribbert H, et al. (February 2003). "Targeting bcl-2 by triplex-forming oligonucleotide--a promising carrier for gene-radiotherapy". Cancer Biotherapy & Radiopharmaceuticals. 18 (1): 17–26. doi:10.1089/108497803321269296. PMID 12667305.
- Sakamoto N, Chastain PD, Parniewski P, Ohshima K, Pandolfo M, Griffith JD, Wells RD (April 1999). "Sticky DNA: self-association properties of long GAA.TTC repeats in R.R.Y triplex structures from Friedreich's ataxia". Molecular Cell. 3 (4): 465–75. doi:10.1016/s1097-2765(00)80474-8. PMID 10230399.
- Bacolla A, Wells RD (April 2009). "Non-B DNA conformations as determinants of mutagenesis and human disease". Molecular Carcinogenesis. 48 (4): 273–85. doi:10.1002/mc.20507. PMID 19306308. S2CID 5493647.
- Kaushik Tiwari M, Adaku N, Peart N, Rogers FA (September 2016). "Triplex structures induce DNA double strand breaks via replication fork collapse in NER deficient cells". Nucleic Acids Research. 44 (16): 7742–54. doi:10.1093/nar/gkw515. PMC 5027492. PMID 27298253.
- Jain A, Wang G, Vasquez KM (August 2008). "DNA triple helices: biological consequences and therapeutic potential". Biochimie. 90 (8): 1117–30. doi:10.1016/j.biochi.2008.02.011. PMC 2586808. PMID 18331847.
- Hansen ME, Bentin T, Nielsen PE (July 2009). "High-affinity triplex targeting of double stranded DNA using chemically modified peptide nucleic acid oligomers". Nucleic Acids Research. 37 (13): 4498–507. doi:10.1093/nar/gkp437. PMC 2715256. PMID 19474349.
- Ricciardi AS, McNeer NA, Anandalingam KK, Saltzman WM, Glazer PM (2014). "Targeted genome modification via triple helix formation". In Wajapeyee N (ed.). Cancer Genomics and Proteomics. Methods in Molecular Biology. 1176. New York, NY: Springer New York. pp. 89–106. doi:10.1007/978-1-4939-0992-6_8. ISBN 978-1-4939-0991-9. PMC 5111905. PMID 25030921.
- Rogers FA, Vasquez KM, Egholm M, Glazer PM (December 2002). "Site-directed recombination via bifunctional PNA-DNA conjugates". Proceedings of the National Academy of Sciences of the United States of America. 99 (26): 16695–700. Bibcode:2002PNAS...9916695R. doi:10.1073/pnas.262556899. PMC 139206. PMID 12461167.
- Montazersaheb S, Hejazi MS, Nozad Charoudeh H (November 2018). "Potential of Peptide Nucleic Acids in Future Therapeutic Applications". Advanced Pharmaceutical Bulletin. 8 (4): 551–563. doi:10.15171/apb.2018.064. PMC 6311635. PMID 30607328.
- Firulli AB, Maibenco DC, Kinniburgh AJ (April 1994). "Triplex forming ability of a c-myc promoter element predicts promoter strength". Archives of Biochemistry and Biophysics. 310 (1): 236–42. doi:10.1006/abbi.1994.1162. PMID 8161210.
- Zain R, Sun JS (May 2003). "Do natural DNA triple-helical structures occur and function in vivo?". Cellular and Molecular Life Sciences. 60 (5): 862–70. doi:10.1007/s00018-003-3046-3. PMID 12827276. S2CID 6227195.
- Kato M, Shimizu N (October 1992). "Effect of the potential triplex DNA region on the in vitro expression of bacterial beta-lactamase gene in superhelical recombinant plasmids". Journal of Biochemistry. 112 (4): 492–4. doi:10.1093/oxfordjournals.jbchem.a123927. PMID 1491004.
- Potaman VN, Ussery DW, Sinden RR (June 1996). "Formation of a combined H-DNA/open TATA box structure in the promoter sequence of the human Na,K-ATPase alpha2 gene". The Journal of Biological Chemistry. 271 (23): 13441–7. doi:10.1074/jbc.271.23.13441. PMID 8662935. S2CID 46550975.
- Seidman MM, Glazer PM (August 2003). "The potential for gene repair via triple helix formation". The Journal of Clinical Investigation. 112 (4): 487–94. doi:10.1172/JCI19552. PMC 171401. PMID 12925687.
- Rapozzi V, Cogoi S, Spessotto P, Risso A, Bonora GM, Quadrifoglio F, Xodo LE (January 2002). "Antigene effect in K562 cells of a PEG-conjugated triplex-forming oligonucleotide targeted to the bcr/abl oncogene". Biochemistry. 41 (2): 502–10. doi:10.1021/bi011314h. PMID 11781088.
- Bertucat G, Lavery R, Prévost C (December 1998). "A model for parallel triple helix formation by RecA: single-single association with a homologous duplex via the minor groove". Journal of Biomolecular Structure & Dynamics. 16 (3): 535–46. doi:10.1080/07391102.1998.10508268. PMID 10052612.
- Chen J, Tang Q, Guo S, Lu C, Le S, Yan J (September 2017). "Parallel triplex structure formed between stretched single-stranded DNA and homologous duplex DNA". Nucleic Acids Research. 45 (17): 10032–10041. doi:10.1093/nar/gkx628. PMC 5622322. PMID 28973442.
- Camerini-Otero RD, Hsieh P (April 1993). "Parallel DNA triplexes, homologous recombination, and other homology-dependent DNA interactions". Cell. 73 (2): 217–23. doi:10.1016/0092-8674(93)90224-e. PMID 8477443. S2CID 34585948.
- Bertucat G, Lavery R, Prévost C (September 1999). "A molecular model for RecA-promoted strand exchange via parallel triple-stranded helices". Biophysical Journal. 77 (3): 1562–76. Bibcode:1999BpJ....77.1562B. doi:10.1016/S0006-3495(99)77004-9. PMC 1300444. PMID 10465767.
- Yang H, Zhou C, Dhar A, Pavletich NP (October 2020). "Mechanism of strand exchange from RecA-DNA synaptic and D-loop structures". Nature. 586 (7831): 801–806. doi:10.1038/s41586-020-2820-9. PMID 33057191. S2CID 222349650.
- McKinney JA, Wang G, Mukherjee A, Christensen L, Subramanian SH, Zhao J, Vasquez KM (January 2020). "Distinct DNA repair pathways cause genomic instability at alternative DNA structures". Nature Communications. 11 (1): 236. Bibcode:2020NatCo..11..236M. doi:10.1038/s41467-019-13878-9. PMC 6957503. PMID 31932649.
- Wang G, Vasquez KM (July 2014). "Impact of alternative DNA structures on DNA damage, DNA repair, and genetic instability". DNA Repair. 19: 143–51. doi:10.1016/j.dnarep.2014.03.017. PMC 4216180. PMID 24767258.
- Raghavan SC, Chastain P, Lee JS, Hegde BG, Houston S, Langen R, et al. (June 2005). "Evidence for a triplex DNA conformation at the bcl-2 major breakpoint region of the t(14;18) translocation". The Journal of Biological Chemistry. 280 (24): 22749–60. doi:10.1074/jbc.M502952200. PMID 15840562.
- Wang G, Vasquez KM (September 2004). "Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells". Proceedings of the National Academy of Sciences of the United States of America. 101 (37): 13448–53. Bibcode:2004PNAS..10113448W. doi:10.1073/pnas.0405116101. PMC 518777. PMID 15342911.
- Vetcher AA, Napierala M, Iyer RR, Chastain PD, Griffith JD, Wells RD (October 2002). "Sticky DNA, a long GAA.GAA.TTC triplex that is formed intramolecularly, in the sequence of intron 1 of the frataxin gene". The Journal of Biological Chemistry. 277 (42): 39217–27. doi:10.1074/jbc.M205209200. PMID 12161437.
- Wang G, Vasquez KM (January 2017). "Effects of Replication and Transcription on DNA Structure-Related Genetic Instability". Genes. 8 (1): 17. doi:10.3390/genes8010017. PMC 5295012. PMID 28067787.
- Zhao J, Wang G, Del Mundo IM, McKinney JA, Lu X, Bacolla A, et al. (January 2018). "Distinct Mechanisms of Nuclease-Directed DNA-Structure-Induced Genetic Instability in Cancer Genomes". Cell Reports. 22 (5): 1200–1210. doi:10.1016/j.celrep.2018.01.014. PMC 6011834. PMID 29386108.
- Belotserkovskii BP, De Silva E, Tornaletti S, Wang G, Vasquez KM, Hanawalt PC (November 2007). "A triplex-forming sequence from the human c-MYC promoter interferes with DNA transcription". The Journal of Biological Chemistry. 282 (44): 32433–41. doi:10.1074/jbc.M704618200. PMID 17785457. S2CID 24211097.
- Pandey S, Ogloblina AM, Belotserkovskii BP, Dolinnaya NG, Yakubovskaya MG, Mirkin SM, Hanawalt PC (August 2015). "Transcription blockage by stable H-DNA analogs in vitro". Nucleic Acids Research. 43 (14): 6994–7004. doi:10.1093/nar/gkv622. PMC 4538819. PMID 26101261.
- Del Mundo IM, Zewail-Foote M, Kerwin SM, Vasquez KM (May 2017). "Alternative DNA structure formation in the mutagenic human c-MYC promoter". Nucleic Acids Research. 45 (8): 4929–4943. doi:10.1093/nar/gkx100. PMC 5416782. PMID 28334873.
- McNeer NA, Schleifman EB, Cuthbert A, Brehm M, Jackson A, Cheng C, et al. (June 2013). "Systemic delivery of triplex-forming PNA and donor DNA by nanoparticles mediates site-specific genome editing of human hematopoietic cells in vivo". Gene Therapy. 20 (6): 658–69. doi:10.1038/gt.2012.82. PMC 3713483. PMID 23076379.
- Hnedzko D, Cheruiyot SK, Rozners E (September 2014). "Using triple-helix-forming Peptide nucleic acids for sequence-selective recognition of double-stranded RNA". Current Protocols in Nucleic Acid Chemistry. 58: 4.60.1–23. doi:10.1002/0471142700.nc0460s58. ISSN 1934-9270. PMC 4174339. PMID 25199637.
- Schleifman EB, Bindra R, Leif J, del Campo J, Rogers FA, Uchil P, et al. (September 2011). "Targeted disruption of the CCR5 gene in human hematopoietic stem cells stimulated by peptide nucleic acids". Chemistry & Biology. 18 (9): 1189–98. doi:10.1016/j.chembiol.2011.07.010. PMC 3183429. PMID 21944757.
- McNeer NA, Anandalingam K, Fields RJ, Caputo C, Kopic S, Gupta A, et al. (April 2015). "Nanoparticles that deliver triplex-forming peptide nucleic acid molecules correct F508del CFTR in airway epithelium". Nature Communications. 6 (1): 6952. Bibcode:2015NatCo...6.6952M. doi:10.1038/ncomms7952. PMC 4480796. PMID 25914116.
- Lukacs GL, Verkman AS (February 2012). "CFTR: folding, misfolding and correcting the ΔF508 conformational defect". Trends in Molecular Medicine. 18 (2): 81–91. doi:10.1016/j.molmed.2011.10.003. PMC 3643519. PMID 22138491.
- Pauling L, Corey RB (February 1953). "A Proposed Structure For The Nucleic Acids". Proceedings of the National Academy of Sciences of the United States of America. 39 (2): 84–97. Bibcode:1953PNAS...39...84P. doi:10.1073/pnas.39.2.84. PMC 1063734. PMID 16578429.
- Pauling L, Corey RB (February 1953). "Structure of the nucleic acids". Nature. 171 (4347): 346. Bibcode:1953Natur.171..346P. doi:10.1038/171346a0. PMID 13036888. S2CID 4151877.
- Fraser RD (March 2004). "The structure of deoxyribose nucleic acid". Journal of Structural Biology. 145 (3): 184–5. doi:10.1016/j.jsb.2004.01.001. PMID 14997898.
- Felsenfeld G, Davies DR, Rich A (April 1957). "Formation of a three-stranded polynucleotide molecule". Journal of the American Chemical Society. 79 (8): 2023–4. doi:10.1021/ja01565a074.
- Hanvey JC, Shimizu M, Wells RD (September 1988). "Intramolecular DNA triplexes in supercoiled plasmids". Proceedings of the National Academy of Sciences of the United States of America. 85 (17): 6292–6. Bibcode:1988PNAS...85.6292H. doi:10.1073/pnas.85.17.6292. PMC 281955. PMID 3413097.
- Mirkin SM, Lyamichev VI, Drushlyak KN, Dobrynin VN, Filippov SA, Frank-Kamenetskii MD (December 1987). "DNA H form requires a homopurine-homopyrimidine mirror repeat". Nature. 330 (6147): 495–7. Bibcode:1987Natur.330..495M. doi:10.1038/330495a0. PMID 2825028. S2CID 4360764.
Further reading
- Rich A (December 1993). "DNA comes in many forms". Gene. 135 (1–2): 99–109. doi:10.1016/0378-1119(93)90054-7. PMID 8276285.
- Soyfer VN, Potaman VN (1995). Triple-Helical Nucleic Acids. New York: Springer. ISBN 978-0-387-94495-1.
- Mills M, Arimondo PB, Lacroix L, Garestier T, Hélène C, Klump H, Mergny JL (September 1999). "Energetics of strand-displacement reactions in triple helices: a spectroscopic study". Journal of Molecular Biology. 291 (5): 1035–54. doi:10.1006/jmbi.1999.3014. PMID 10518941.
- Watson JD, Crick FH (April 1953). "Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid". Nature. 171 (4356): 737–8. Bibcode:1953Natur.171..737W. doi:10.1038/171737a0. PMID 13054692. S2CID 4253007.