Transition metal phosphinimide complexes
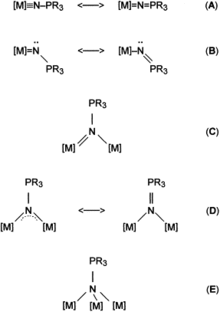
Transition metal phosphinimide complexes are metal complexes that contain phosphinimide ligands of the general formula NPR3− (R = organic substituent). Several coordination modes have been observed, including terminal and various bridging geometries.[1] In the terminal bonding mode the M-N=P core is usually linear but some are quite bent.[1][2] The preferred coordination type varies with the oxidation state and coligands on the metal and the steric and electronic properties of the R groups on phosphorus.[3] Many transition metal phosphinimide complexes have been well-developed and, more recently, main group phosphinimide complexes have been synthesized.[1]
Complexes of Ti, Zr, V, Ta
Complexes of Phosphinimide are generally prepared by two routes.[2] For highly electrophilic metal chlorides, the silyl derivative is convenient since is generates volatile trimethylsilyl chloride:
- R3PNSiMe3 + LnMCl → R3PN-MLn + ClSiMe3
The synthesis of CpTi(NPR3)Cl2 is prepared by this route.
More common are salt-elimination reactions:
- R3PNLi + LnMCl → R3PN-MLn + LiCl
Phosphinimide polyethylene catalysts
Phosphinimide ligands have shown promise in the area of ethylene polymerization. In terms of homogeneous catalysts, this field has been dominated by metallocene-based catalysts inspired by the Kaminsky catalyst in 1976.[4] Initially phosphinimide ligands were suggested for polyethylene synthesis due to the fact they have similar steric and electronic properties to metallocene polyethylene catalysts.[5]
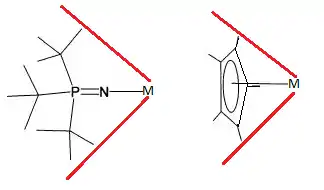
In most respects the steric and electronic properties, of phosphinimides and Cylclopentadienyl are comparable ligands.[5][6][7] Metal bound t-Bu3PN− has a cone angle of 87° vs 83 for cylclopentadienyl.[5] Compared to Cp, the bulky substituents of the phosphinimide ligand are more distant from the metal, which increase the exposure of the metal centre to substrate.[5] The less sterically crowded metal centre appears to be particularly susceptible to deactivation however.[5][6][7]
The precatalyst are prepared by alkylation and arylation of the phosphinimide complexes is possible through alkyllithium or Grignard reagents, giving products such as CpTi(NPR3)Me2. The zirconium complexes (R3PN)2ZrCl2 can be alkylated or arylated through simple substitution.[2] These organoTi and organoZr complexes are activated by treatment with MAO[4] and B(C6F5)3 as a cocatalyst to activate polymerization through methyl abstraction.[7] The phosphinimide catalyst is thought to be homogeneous and single sited. It therefore produces reactivity comparable to metallocene catalysts[5][7] which are also believed to be homogeneous, single sited catalysts. The catalytic process is assumed to proceed in much of the same way as metallocene based catalysts, as the chemistry is thought to occur primarily with the metal centre and not through the bulky ligands.[6]
References
- Dehnicke, Kurt; Krieger, M.; Massa, W. (1999). "Phosphoraneiminato Complexes of Transition metals". Coordination Chemistry Reviews. 182: 19–65. doi:10.1016/S0010-8545(97)90055-2.
- Stephan, Douglas W. (2005). "The Road to Early-Transition-Metal Phosphinimide Olefin Polymerization Catalysts". Organometallics. 24 (11): 2548–2560. doi:10.1021/om050096b.
- Stephan, Douglas (2006). "Sterically Demanding Phosphinimides: Ligands for Unique Main Group and Transition Metal Chemistry". Advances in Organometallic Chemistry. 54: 267–291. doi:10.1016/S0065-3055(05)54006-1.
- Reddy, S.; Sivaram, S. (1995). "Homogeneous Metallocene-Methylaluminoxane Catalyst Systems for Ethylene Polymerization". Prog. Polym. Sci. 20: 309–367.
- Stephan, D.; Stewart, J.; et al. (2003). "An Approach to Catalyst Design: Cyclopentadienyl-Titanium Phosphinimide Complexes in Ethylene Polymerization". Organometallics. 22: 1937–47. doi:10.1021/om020954t.
- Voth, P.; Fraser, C.; et al. (2006). "Functionalizing Titanium-Phosphinimide Complexes". Organometallics. 25: 4779–86. doi:10.1021/om060565p.
- Stephan, D. (2002). "Alcan Award Lecture: From Academic Research to Industrial Applications and Back Again". Can. J. Chem. 80: 125–32. doi:10.1139/V01-203.