Supercritical fluid extraction
Supercritical fluid extraction (SFE) is the process of separating one component (the extractant) from another (the matrix) using supercritical fluids as the extracting solvent. Extraction is usually from a solid matrix, but can also be from liquids. SFE can be used as a sample preparation step for analytical purposes, or on a larger scale to either strip unwanted material from a product (e.g. decaffeination) or collect a desired product (e.g. essential oils). These essential oils can include limonene and other straight solvents. Carbon dioxide (CO2) is the most used supercritical fluid, sometimes modified by co-solvents such as ethanol or methanol. Extraction conditions for supercritical carbon dioxide are above the critical temperature of 31 °C and critical pressure of 74 bar. Addition of modifiers may slightly alter this. The discussion below will mainly refer to extraction with CO2, except where specified.
Advantages
Selectivity
The properties of the supercritical fluid can be altered by varying the pressure and temperature, allowing selective extraction. For example, volatile oils can be extracted from a plant with low pressures (100 bar), whereas liquid extraction would also remove lipids. Lipids can be removed using pure CO2 at higher pressures, and then phospholipids can be removed by adding ethanol to the solvent.[1] The same principle can be used to extract polyphenols and unsaturated fatty acids separately from wine wastes.[2]
Speed
Extraction is a diffusion-based process, in which the solvent is required to diffuse into the matrix and the extracted material to diffuse out of the matrix into the solvent. Diffusivities are much faster in supercritical fluids than in liquids, and therefore extraction can occur faster. In addition, due to the lack of surface tension and negligible viscosities compared to liquids, the solvent can penetrate more into the matrix inaccessible to liquids. An extraction using an organic liquid may take several hours, whereas supercritical fluid extraction can be completed in 10 to 60 minutes.[3]
Limitations
The requirement for high pressures increases the cost compared to conventional liquid extraction, so SFE will only be used where there are significant advantages. Carbon dioxide itself is non-polar, and has somewhat limited dissolving power, so cannot always be used as a solvent on its own, particularly for polar solutes. The use of modifiers increases the range of materials which can be extracted. Food grade modifiers such as ethanol can often be used, and can also help in the collection of the extracted material, but reduces some of the benefits of using a solvent which is gaseous at room temperature.
Procedure
The system must contain a pump for the CO2, a pressure cell to contain the sample, a means of maintaining pressure in the system and a collecting vessel. The liquid is pumped to a heating zone, where it is heated to supercritical conditions. It then passes into the extraction vessel, where it rapidly diffuses into the solid matrix and dissolves the material to be extracted. The dissolved material is swept from the extraction cell into a separator at lower pressure, and the extracted material settles out. The CO2 can then be cooled, re-compressed and recycled, or discharged to atmosphere.
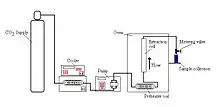
Pumps
Carbon dioxide (CO
2) is usually pumped as a liquid, usually below 5 °C (41 °F) and a pressure of about 50 bar. The solvent is pumped as a liquid as it is then almost incompressible; if it were pumped as a supercritical fluid, much of the pump stroke would be "used up" in compressing the fluid, rather than pumping it. For small scale extractions (up to a few grams / minute), reciprocating CO
2 pumps or syringe pumps are often used. For larger scale extractions, diaphragm pumps are most common. The pump heads will usually require cooling, and the CO2 will also be cooled before entering the pump.
Pressure vessels
Pressure vessels can range from simple tubing to more sophisticated purpose built vessels with quick release fittings. The pressure requirement is at least 74 bar, and most extractions are conducted at under 350 bar. However, sometimes higher pressures will be needed, such as extraction of vegetable oils, where pressures of 800 bar are sometimes required for complete miscibility of the two phases.[4]
The vessel must be equipped with a means of heating. It can be placed inside an oven for small vessels, or an oil or electrically heated jacket for larger vessels. Care must be taken if rubber seals are used on the vessel, as the supercritical carbon dioxide may dissolve in the rubber, causing swelling, and the rubber will rupture on depressurization.
Pressure maintenance
The pressure in the system must be maintained from the pump right through the pressure vessel. In smaller systems (up to about 10 mL / min) a simple restrictor can be used. This can be either a capillary tube cut to length, or a needle valve which can be adjusted to maintain pressure at different flow rates. In larger systems a back pressure regulator will be used, which maintains pressure upstream of the regulator by means of a spring, compressed air, or electronically driven valve. Whichever is used, heating must be supplied, as the adiabatic expansion of the CO2 results in significant cooling. This is problematic if water or other extracted material is present in the sample, as this may freeze in the restrictor or valve and cause blockages.
Collection
The supercritical solvent is passed into a vessel at lower pressure than the extraction vessel. The density, and hence dissolving power, of supercritical fluids varies sharply with pressure, and hence the solubility in the lower density CO2 is much lower, and the material precipitates for collection. It is possible to fractionate the dissolved material using a series of vessels at reducing pressure. The CO2 can be recycled or depressurized to atmospheric pressure and vented. For analytical SFE, the pressure is usually dropped to atmospheric, and the now gaseous carbon dioxide bubbled through a solvent to trap the precipitated components.
Heating and cooling
This is an important aspect. The fluid is cooled before pumping to maintain liquid conditions, then heated after pressurization. As the fluid is expanded into the separator, heat must be provided to prevent excessive cooling. For small scale extractions, such as for analytical purposes, it is usually sufficient to pre-heat the fluid in a length of tubing inside the oven containing the extraction cell. The restrictor can be electrically heated, or even heated with a hairdryer. For larger systems, the energy required during each stage of the process can be calculated using the thermodynamic properties of the supercritical fluid.[5]
Simple model of SFE
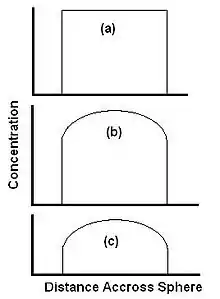
There are two essential steps to SFE, transport (by diffusion or otherwise) of the solid particles to the surface, and dissolution in the supercritical fluid. Other factors, such as diffusion into the particle by the SF and reversible release such as desorption from an active site are sometimes significant, but not dealt with in detail here. Figure 2 shows the stages during extraction from a spherical particle where at the start of the extraction the level of extractant is equal across the whole sphere (Fig. 2a). As extraction commences, material is initially extracted from the edge of the sphere, and the concentration in the center is unchanged (Fig 2b). As the extraction progresses, the concentration in the center of the sphere drops as the extractant diffuses towards the edge of the sphere (Figure 2c).[6]
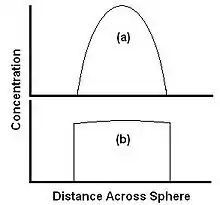
The relative rates of diffusion and dissolution are illustrated by two extreme cases in Figure 3. Figure 3a shows a case where dissolution is fast relative to diffusion. The material is carried away from the edge faster than it can diffuse from the center, so the concentration at the edge drops to zero. The material is carried away as fast as it arrives at the surface, and the extraction is completely diffusion limited. Here the rate of extraction can be increased by increasing diffusion rate, for example raising the temperature, but not by increasing the flow rate of the solvent. Figure 3b shows a case where solubility is low relative to diffusion. The extractant is able to diffuse to the edge faster than it can be carried away by the solvent, and the concentration profile is flat. In this case, the extraction rate can be increased by increasing the rate of dissolution, for example by increasing flow rate of the solvent.
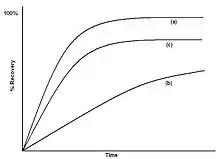
The extraction curve of % recovery against time can be used to elucidate the type of extraction occurring. Figure 4(a) shows a typical diffusion controlled curve. The extraction is initially rapid, until the concentration at the surface drops to zero, and the rate then becomes much slower. The % extracted eventually approaches 100%. Figure 4(b) shows a curve for a solubility limited extraction. The extraction rate is almost constant, and only flattens off towards the end of the extraction. Figure 4(c) shows a curve where there are significant matrix effects, where there is some sort of reversible interaction with the matrix, such as desorption from an active site. The recovery flattens off, and if the 100% value is not known, then it is hard to tell that extraction is less than complete.
Optimization
The optimum will depend on the purpose of the extraction. For an analytical extraction to determine, say, antioxidant content of a polymer, then the essential factors are complete extraction in the shortest time. However, for production of an essential oil extract from a plant, then quantity of CO2 used will be a significant cost, and "complete" extraction not required, a yield of 70 - 80% perhaps being sufficient to provide economic returns. In another case, the selectivity may be more important, and a reduced rate of extraction will be preferable if it provides greater discrimination. Therefore, few comments can be made which are universally applicable. However, some general principles are outlined below.
Maximizing diffusion
This can be achieved by increasing the temperature, swelling the matrix, or reducing the particle size. Matrix swelling can sometimes be increased by increasing the pressure of the solvent, and by adding modifiers to the solvent. Some polymers and elastomers in particular are swelled dramatically by CO2, with diffusion being increased by several orders of magnitude in some cases.[7]
Maximizing solubility
Generally, higher pressure will increase solubility. The effect of temperature is less certain, as close to the critical point, increasing the temperature causes decreases in density, and hence dissolving power. At pressures well above the critical pressure, solubility is likely to increase with temperature.[8] Addition of low levels of modifiers (sometimes called entrainers), such as methanol and ethanol, can also significantly increase solubility, particularly of more polar compounds.
Optimizing flow rate
The flow rate of supercritical carbon dioxide should be measured in terms of mass flow rather than by volume because the density of the CO
2 changes according to the temperature both before entering the pump heads and during compression. Coriolis flow meters are best used to achieve such flow confirmation. To maximize the rate of extraction, the flow rate should be high enough for the extraction to be completely diffusion limited (but this will be very wasteful of solvent). However, to minimize the amount of solvent used, the extraction should be completely solubility limited (which will take a very long time). Flow rate must therefore be determined depending on the competing factors of time and solvent costs, and also capital costs of pumps, heaters and heat exchangers. The optimum flow rate will probably be somewhere in the region where both solubility and diffusion are significant factors.
References
- Tanaka, Y.; Takeshi, O (2004). "Extraction of Phospholipids from salmon roe with supercritical carbon dioxide and an entrainer". Journal of Oleo Science. 53 (9): 417–424. doi:10.5650/jos.53.417. Retrieved 2007-12-05.
- Aizpurua-Olaizola, Oier; Ormazabal, Markel; Vallejo, Asier; Olivares, Maitane; Navarro, Patricia; Etxebarria, Nestor; Usobiaga, Aresatz (2015-01-01). "Optimization of Supercritical Fluid Consecutive Extractions of Fatty Acids and Polyphenols from Vitis Vinifera Grape Wastes". Journal of Food Science. 80 (1): E101–E107. doi:10.1111/1750-3841.12715. ISSN 1750-3841. PMID 25471637.
- Skoog (2007) [1998]. "29". Principles of Instrumental Analysis. David Harris. p. 863. ISBN 978-0-495-01201-6.
- King, Jerry W. (2002). "34, Supercritical Fluid Technology for Lipid Extraction, Fractionation and Reactions" (PDF). In Tsung Min Kuo and Harold Gardner (ed.). Lipid Biotechnology. New York: Marcel Dekker Inc. pp. 663–687.
- "Calculation of Density, Enthalpy and Entropy for Supercritical Carbon Dioxide with Examples". Archived from the original on 2008-05-05. Retrieved 2007-12-17.
- Clifford, Tony (1999). Fundamentals of Supercritical Fluids. Oxford: Oxford Science Publications. ISBN 978-0-19-850137-4.
- Vandenburg, H. J.; Clifford, Anthony A.; et al. (1997). "Analytical Extraction of Additives from Polymers". Analyst. 122 (9): 101R–115R. doi:10.1039/a704052k.
- "Supercritical Fluid Extraction, Density Considerations". Retrieved 2008-01-04.
Further reading
- McHugh, Mark A.; Krukonis, Val J. (1994). Supercritical Fluid Extraction - Principles and Practice. Butterworth Heinemann series in chemical engineering (2nd ed.). Butterworth Heinemann. ISBN 978-0-7506-9244-1.
- Taylor, Larry T (1996). Supercritical Fluid Extraction. Techniques in analytical chemistry. John Wiley and Sons, Inc. ISBN 978-0-471-11990-6.