Stereoelectronic effect
In chemistry, primarily organic and computational chemistry, a stereoelectronic effect[1] is an effect on molecular geometry, reactivity, or physical properties due to spatial relationships in the molecules' electronic structure, in particular the interaction between atomic and/or molecular orbitals.[2] Phrased differently, stereoelectronic effects can also be defined as the geometric constraints placed on the ground and/or transition states of molecules that arise from considerations of orbital overlap.[3] Thus, a stereoelectronic effect explains a particular molecular property or reactivity by invoking stabilizing or destabilizing interactions that depend on the relative orientations of electrons (bonding or non-bonding) in space.[4]
Founded on a few general principles that govern how orbitals interact, the stereoelectronic effect, along with the steric effect, inductive effect, solvent effect, mesomeric effect, and aromaticity, is an important type of explanation for observed patterns of selectivity, reactivity and stability in organic chemistry. In spite of the relatively straightforward premises, stereoelectronic effects often provide explanations for counterintuitive or surprising observations. As a result, stereoelectronic factors are now commonly considered and exploited in the development of new organic methodology and in the synthesis of complex targets. The scrutiny of stereoelectronic effects has also entered the realms of biochemistry and pharmaceutical chemistry in recent years.
A stereoelectronic effect generally involves a stabilizing donor–acceptor (i.e., filled-empty, 2-electron 2-orbital) interaction. The donor is usually a higher bonding or nonbonding orbital and the acceptor is often a low-lying antibonding orbital as shown in the scheme below. Whenever possible, if this stereoelectronic effect is to be favored, the donor–acceptor orbitals should have (1) a small energy gap and (2) be geometrically well disposed for interaction. In particular, this means that the shapes of the donor and acceptor orbitals (including π or σ symmetry and size of the interacting lobes) must be well-matched for interaction; an antiperiplanar orientation is especially favorable. Some authors require stereoelectronic effects to be stabilizing.[1] However, destabilizing donor-donor (i.e., filled-filled, 4-electron 2-orbital) interactions are occasionally invoked and are also sometimes referred to as stereoelectronic effects, although such effects are difficult to distinguish from generic steric repulsion.[3][5]
Important phenomena in which stereoelectronic effects and orbital alignment may play an important role (or may even be dominant) include the anomeric effect and hyperconjugation.[5]
The term stereoelectronic effect should not be misused to refer to a simple combination of steric and electronic effects.
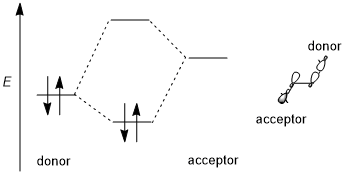
Trend of different orbitals
Take the simplest CH2X–CH3 system as an example; the donor orbital is σ(C–H) orbital and the acceptor is σ*(C–X). When moving from fluorine to chlorine, then to bromine, the electronegativity of the halogen and the energy level of the σ*(C–X) orbitals decreases.[6] Consequently, the general trend of acceptors can be summarized as: π*(C=O)>σ*(C–Hal)>σ*(C–O)>σ*(C–N)>σ*(C–C), σ*(C–H). For donating orbitals, the nonbonding orbitals, or the lone pairs, are generally more effective than bonding orbitals due to the high energy levels. Also, different from acceptors, donor orbitals require less polarized bonds. Thus, the general trends for donor orbitals would be: n(N)>n(O)>σ(C–C), σ(C–H)>σ(C–N)>σ(C–O)>σ(C–S)>σ(C–Hal).[5]
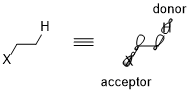
Stereoelectronic effect can be directional in specific cases. The radius of sulfur is much larger than the radius of carbon and oxygen. Thus the differences in C–S bond distances generate a much amplified difference in the two stereoelectronic effects in 1,3-dithiane (σ(C–H) → σ*(C–S)) than in 1,3-dioxane(σ(C–H) → σ*(C–O)).[6] The differences between C–C and C–S bonds shown below causes a significant difference in the distances between C–S and two C–H bonds. The shorter the difference is, the better the interaction and the stronger the stereoelectronic effect.[6]

Influence on stability
If there is an electropositive substituent (e.g. –SiR3, –SnR3, –HgR, etc.) at the β-position of carbocation, the positive charge could be stabilized which is also due largely to the stereoelectronic effect (illustrated below using –SiR3 as an example). The orientation of the two interacting orbitals can have a significant effect on the stabilization effect (σ(C–Si) → empty p orbital), where antiperiplanar (180°) > perpendicular (90°) > syn (0°).[7]

Influence on conformation
Gauche effect
One structural consequence of acyclic systems due to the stereoelectronic effect is the gauche effect.[8] In 1,2-difluoroethane, despite the steric clash, the preferred conformation is the gauche one because σ(C–H) is a good donor and σ*(C–F) is a good acceptor and the stereoelectronic effect (σ(C–H) → σ*(C–F)) requires the energy minimum to be gauche instead of anti.[9]

This gauche effect has a profound effect in bio-chemical research. In (2S,4R)-4-hydroxyproline fragment, the gauche interaction favors one the conformer that can bind selectively to the active site of pVHL, a domain in collagen, one of the most abundant animal protein structures and can lead to proteasomal degradation of HIF-α subunit.[10]

Special effects of fluorine substituent
Stereoelectronic effects can have a significant influence in pharmaceutical research. Generally, the substitution of hydrogen by fluorine could be regarded as a way to tune both the hydrophobicity and the metabolic stability of a drug candidate. Moreover, it can have a profound influence on conformations, often due to stereoelectronic effects, in addition to normal steric effects resulting from the larger size of the fluorine atom. For instance, the ground state geometries of anisole (methoxybenzene) and (trifluoromethoxy)benzene differ dramatically. In anisole, the methyl group prefers to be coplanar with the phenyl group, while (trifluoromethoxy)benzene favors a geometry in which the [C(aryl)–C(aryl)–O–C(F3)] dihedral angle is around 90°. In other words, the O–CF3 bond is perpendicular to the plane of the phenyl group.[11]
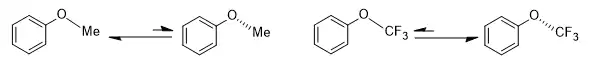
Further studies illustrate that even for only one or two hydrogen atoms in a methyl group being replaced by a fluorine atom, the distortion in the structure can also be significant, with the [C(aryl)–C(aryl)–O–C(H2F)] dihedral angle in the energy minimized structure being around 24° and the [C(aryl)–C(aryl)–O–C(HF2)] dihedral angle 33°.[11]
Influence on reaction selectivity
Reductive cyclizations
Although the energy difference between coplanar anisole and its isomer is quite large, the rotation between the O–CH3 bond becomes favorable when the electronic properties of methoxy group on aromatic rings need to be altered to stabilize an unusual intermediate or a transition state. In the following reaction, the regioselectivity could be rationalized as the out-of-plane rotation of the O–C bond which changes the methoxy group from an in-plane donor group to an out-of-plane acceptor group.[12]

The intermediate of the above reaction is the di-anion and the stereoelectronic effect that stabilizes this intermediate over the other one is the fact that the anionic charge at the para position could delocalize to the oxygen atom via orbital interaction: π(benzene) → σ*(O–CH3).[12]

Hydrogenation
Even remote substituents on the benzene ring can affect the electron density on the aromatic ring and in turn influence the selectivity. In the hydrogenation of ketones using CBS catalysts, the ketone coordinates to the boron atom with the lone pair on the oxygen atom. In the following example, the inductive influence of the substituents can lead to differentiation of the two sp2 lone pairs on the oxygen atom.[13]
The relevant stereoelectronic interaction in the starting material is the nO → σ*(Ccarbonyl–Caryl) interaction. The electron-withdrawing substituent on the benzene ring depletes the electron density on the aromatic ring and thus makes the σ*(Ccarbonyl–Caryl(nitro)) orbital a better acceptor than σ*(Ccarbonyl–Caryl(methoxy)). These two stereoelectronic interactions use different lone pairs on the oxygen atom (the one antiperiplanar to the σ* in question for each), leading to lone pairs with different electron densities. In particular, the enhanced depletion of electron density from the lone pair antiperiplanar to the 4-nitrophenyl group leads to weakened ability for that lone pair to coordinate to boron. This in turn results in the lone pair antiperiplanar to the 4-methoxyphenyl binding preferentially to the catalyst, leading to well-defined facial selectivity. Under optimized conditions, the product is formed with excellent levels of enantioselectivity (95% ee).[13]

Influence on thermodynamics
Influence on equilibrium
The stereoelectronic effect influences the thermodynamics of equilibrium. For example, the following equilibrium could be achieved via a cascade of pericyclic reactions.

Despite very similar structures, one of the two isomers is strongly favored over the other because of a stereoelectronic effect. Since the σ*C-C orbital adjacent to the electron-withdrawing carbonyl group is lower in energy and is therefore a better acceptor than the σ*C-C orbital adjacent to the methoxy, the isomer in which the nO(σ) lone pair is able to donate into this lower-energy antibonding orbital will be stabilized (orbital interaction illustrated).[14]
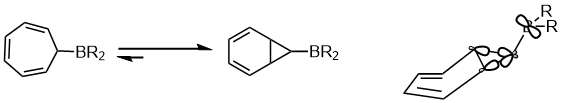
Another example of the preference in the equilibrium within the area of pericyclic reaction is shown below. The stereoelectronic effect that affect the equilibrium is the interaction between the delocalized “banana bonds” and the empty p orbital on the boron atom.[15]
Influence on resonance structures
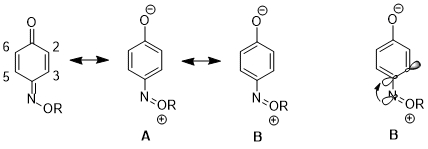
In another case, the stereoelectronic effect can result in an increased contribution of one resonance structure over another, which leads to further consequences in reactivity. For 1,4-benzoquinone monoxime, there are significant differences in the physical properties and reactivities between C2-C3 double bond and C5-C6 double bond. For instance, in the 1H NMR, 3J23 higher than 3J56.[16] The C2-C3 double bond also selectively undergoes Diels–Alder reaction with cyclopentadiene, despite the increased steric hindrance on that side of the molecule.[17] These data illustrate an increased contribution of resonance structure B over structure A. The authors argue that the donation from nN to σ*C4-C3 orbital lengthens the C4–C3 bond (C4 is the carbon bearing the nitrogen substituent), which reduces the p-p overlap between these two atoms. This in turn decreasing the relative importance of structure A which has a double bond between C4 and C3.[18]
Application in asymmetric Diels–Alder reactions
In the asymmetric Diels–Alder reactions, instead of using chiral ligands or chiral auxiliaries to differentiate the side selectivity of the dienolphiles, the differentiation of face selectivity of the dienes (especially for cyclopentadiene derivatives) using stereoelectronic effects have been reported by Woodward since 1955.[19] A systematic research of facial selectivity using substituted cyclopentadiene or permethylcyclopentadiene derivatives have been conducted and the results can be listed as below.[20]

The stereoelectronic effect affecting the outcome of the facial selectivity of the diene in Diels–Alder reaction is the interaction between the σ(C(sp2)–CH3) (when σ(C(sp2)–X) is a better acceptor than a donor) or σ(C(sp2)–X) (when σ(C(sp2)–X) is a better donor than an acceptor) and the σ* orbital of the forming bond between the diene and the dienophile.[20]
If the two geminal substituents are both aromatic rings with different substituents tuning the electron density, the differentiation of the facial selectivity is also facile where the dienophile approaches to the diene anti to the more electron-rich C–C bond where the stereoelectronic effect in this case is similar to the previous one.[21]
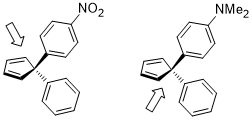
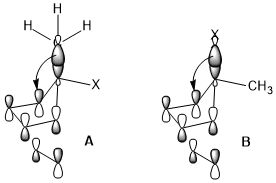
The ring opening of cyclobutene under heating conditions can have two products: inward and outward rotation.

The inward rotation transition state of the second shown below is relatively favored for acceptor R substituents (e.g. NO2) but is especially disfavored by donor R substituents (e.g. NMe2).[22]
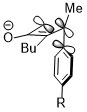
Stereoelectronic effect versus steric clash
Sometimes, stereoelectronic effects can win over extreme steric clash. In a similar cyclobutene ring-opening reaction, the trimethylsilyl group, which is very bulky, still favors the inward rotation. The stereoelectronic effect, which is the interaction shown above when the acceptor orbital is the σ*(Si–CH3), appears to be a more predominant factor in determining the reaction selectivity against the steric hindrance and even wins over the penalty of the disrupted conjugation system of the product due to steric clash.[23]
Furthermore, the acceptor orbitals are not limited to the antibonding orbitals of carbon-heteroatom bonds or the empty orbitals; in the following case, the acceptor orbital is the σ*(B–O) orbital. In the six-membered ring transition state, the stereoelectronic interaction is σ(C–X) → σ*(B–O).[24]

References
- Alabugin, I. V. Stereoelectronic Effects: the Bridge between Structure and Reactivity. John Wiley & Sons Ltd, Chichester, UK, 2016. http://eu.wiley.com/WileyCDA/WileyTitle/productCd-1118906349.html
- Cramer, Christopher J. (1996). "Hyperconjugation as it affects conformational analysis". Journal of Molecular Structure: THEOCHEM. 370 (2–3): 135–146. doi:10.1016/S0166-1280(96)04567-8. ISSN 0166-1280.
- Evans, D. A. (2006). Chemistry 206 Lecture Notes. Cambridge, MA: Harvard University (Unpublished). pp. 1–2 (Lecture 1).
- Pierre., Deslongchamps (1983). Stereoelectronic effects in organic chemistry (1st ed.). Oxford [Oxfordshire]: Pergamon Press. ISBN 0080261841. OCLC 9412829.
- Kirby, A. J. (1996). Stereoelectronic Effects. Oxford: Oxford University Press. ISBN 9780198558934.
- Alabugin, Igor V.; Zeidan, Tarek A. (2002). "Stereoelectronic Effects and General Trends in Hyperconjugative Acceptor Ability of σ Bonds". Journal of the American Chemical Society. 124 (12): 3175–3185. doi:10.1021/ja012633z. ISSN 0002-7863. PMID 11902907.
- Carey, F. A.; Sundberg, R. J. (2007). Advanced Organic Chemistry, Part A: Structure and Mechanism (5th ed.). New York: Springer. ISBN 978-0387448978.
- Hanack, M. (1965). Conformational Theory. New York and London: Academic Press.
- Anslyn, E. V.; Dougherty, D. A. (2004). Modern Physical Organic Chemistry. Sausalito, Calif.: University Science Books. ISBN 1891389319.
- Fujimori, D.G. (2009). "Hypoxia sensing goes gauche". Nat. Chem. Biol. 5 (4): 202–203. doi:10.1038/nchembio0409-202. PMID 19295524.
- Müller, K.; Faeh, C.; Diederich, F. (2007). "Fluorine in Pharmaceuticals: Looking Beyond Intuition". Science. 317 (5846): 1881–1886. Bibcode:2007Sci...317.1881M. doi:10.1126/science.1131943. ISSN 0036-8075. PMID 17901324.
- Peterson, Paul W.; Shevchenko, Nikolay; Alabugin, Igor V. (2013). ""Stereoelectronic Umpolung": Converting a p-Donor into a σ-Acceptor via Electron Injection and a Conformational Change". Organic Letters. 15 (9): 2238–2241. doi:10.1021/ol400813d. ISSN 1523-7060. PMID 23639080.
- Corey, E.J.; Helal, Christopher J. (1995). "Novel electronic effects of remote substituents on the oxazaborolidine-catalyzed enantioselective reduction of ketones". Tetrahedron Letters. 36 (50): 9153–9156. doi:10.1016/0040-4039(95)01961-G. ISSN 0040-4039.
- Venkataraman, Hemalatha; Cha, Jin K. (1989). "The scope and mechanism of rearrangement of 4,6-dialkoxy-2-pyrones". Tetrahedron Letters. 30 (27): 3509–3512. doi:10.1016/S0040-4039(00)99426-7. ISSN 0040-4039.
- Gridnev, Ilya D.; Tok, Oleg L.; Gridneva, Natalya A.; Bubnov, Yuri N.; Schreiner, Peter R. (1998). "Synthesis and Dynamic Properties of Cycloheptatrienyl(dipropyl)borane. Equilibrium with 7-Dipropylborylnorcaradiene". Journal of the American Chemical Society. 120 (5): 1034–1043. doi:10.1021/ja9724699. ISSN 0002-7863.
- Norris, R. K.; Sternhell, S. (1969). "2-Substituted and 2,6-disubstituted 1,4-benzoquinone 4-oximes ("p-nitrosophenols")". Aust. J. Chem. 22 (5): 935–970. doi:10.1071/CH9690935.
- Baldwin, J. E.; Norris, R. K. (1981). "Stereoelectronic control in organic chemistry: addition reactions of some 1,4-benzoquinone 4-(O-methyloximes)". J. Org. Chem. 46 (4): 697–703. doi:10.1021/jo00317a011.
- Perrin, C. L.; Engler, R. E. (1997). "Origin of Apparent Stereoelectronic Effects in Structure and Reactivity of Benzoquinone Monooximes". J. Org. Chem. 62 (3): 687–692. doi:10.1021/jo961386s. PMID 11671465.
- Winstein, S.; Shatavsky, M.; Norton, C.; Woodward, R. B. (1955). "7-Norbornenyl and 7-Norbornyl Cations". J. Am. Chem. Soc. 77 (15): 4183–4184. doi:10.1021/ja01620a078.
- Mehta, Goverdhan; Uma, R. (2000). "Stereoelectronic Control in Diels−Alder Reaction of Dissymmetric 1,3-Dienes". Accounts of Chemical Research. 33 (5): 278–286. doi:10.1021/ar990123s. ISSN 0001-4842. PMID 10813872.
- Halterman, R. L.; McCarthy, B. A.; McEvoy, M. A. (1992). "Stereoelectronic control of facial selectivity in the Diels-Alder cycloaddition of sterically unbiased 5,5-diarylcyclopentadienes". J. Org. Chem. 57 (21): 5585–5589. doi:10.1021/jo00047a009.
- Shindo, M.; Sato, Y.; Shishido, J. (1998). "A highly stereoselective synthesis of tri- and tetrasubstituted olefins via ynolates". Tetrahedron Lett. 39 (27): 4857–4860. doi:10.1016/s0040-4039(98)00921-6.
- Murakami, M.; Hasegawa, M. (2004). "Synthesis and Thermal Ring Opening oftrans-3,4-Disilylcyclobutene". Angew. Chem. Int. Ed. 43 (37): 4874–4876. doi:10.1002/anie.200460144. PMID 15372640.
- Schlapbach, A.; Hoffmann, R. W. (2001). "(E)-α-Sulfonamidocrotylboronates as Reagents for the Stereoselective Homoaldol Synthesis". J. Org. Chem. 66 (2): 323–328. doi:10.1002/1099-0690(200101)2001:2<323::aid-ejoc323>3.0.co;2-a.