Reactions of organocopper reagents
Reactions of organocopper reagents involve species containing copper-carbon bonds acting as nucleophiles in the presence of organic electrophiles. Organocopper reagents are now commonly used in organic synthesis as mild, selective nucleophiles for substitution and conjugate addition reactions.[1]
Since the discovery that copper(I) halides catalyze the conjugate addition of Grignard reagents in 1941,[2] organocopper reagents have emerged as weakly basic, nucleophilic reagents for substitution and addition reactions. The constitution of organocopper compounds depends on their method of preparation and the various kinds of organocopper reagents exhibit different reactivity profiles. As a result, the scope of reactions involving organocopper reagents is extremely broad.
- Organocopper complexes (RCu) are produced when a copper(I) halide and organolithium are combined. In conjunction with Lewis acidic additives such as boron trifluoride etherate, these reagents are used for conjugate addition reactions.[3]
- Lower-order cuprates (R2CuLi, also known as Gilman reagents) result when organocopper complexes are treated with an equivalent of organolithium. Alternatively, they may be formed by the treatment of a copper(I) halide with two equivalents of organolithium. They undergo substitution, conjugate addition, and carbocupration reactions in the presence of the appropriate organic substrates.[4] Mixed Gilman reagents consist of two different R groups, one of which is typically a non-transferable "dummy" group.
- Lower-order cyanocuprates (RCu(CN)Li) are similarly derived from an organolithium compound and copper(I) cyanide; however, intermediate organocopper complexes do not form during this reaction and thus only a single equivalent of organolithium reagent is necessary.[1] Cyanocuprates undergo SN2' substitution in the presence of allyl electrophiles and conjugate addition reactions in the presence of enones.
- Higher-order cyanocuprates (R2Cu(CN)Li2) are formed upon the reaction of two equivalents of organolithium with copper(I) cyanide. These reagents are more reactive towards substitution than the corresponding lower-order cyanocuprates.[5]
Mechanism and Stereochemistry
Substitution Reactions
The mechanism of nucleophilic substitution by lower-order organocuprates depends in a profound way on the structure of the substrate, organocuprate, and reaction conditions. Early evidence suggested that a direct SN2 displacement was occurring;[6] however more recent results suggest that invertive oxidative addition of copper(I) into the carbon-leaving group bond takes place, generating a copper(III) intermediate which then undergoes reductive elimination to generate the coupled product.[7] Both of these mechanisms predict inversion at the electrophilic carbon, which is observed in a number of cases.[8] On the other hand, experiments with radical traps and the observation of racemization during substitution suggest a radical mechanism.[9]
(1)
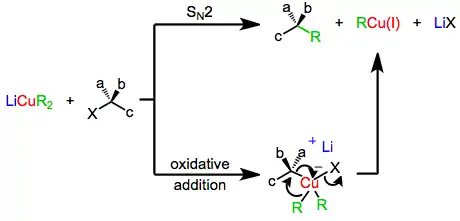
Conjugate Addition Reactions
In 1941, Kharash discovered that Grignard reagents add to cyclohexenone in presence of Cu(I) resulting in 1,4-addition instead of 1,2-addition.[10] This work foreshadowed extensive studies on the conjugate additions to enones with organocuprates. Note that if a Grignard reagent (such as RMgBr) is used, the reaction with an enone would instead proceed through a 1,2-addition. The 1,4-addition mechanism of cuprates to enones goes through the nucleophilic addition of the Cu(I) species at the beta-carbon of the alkene to form a Cu(III) intermediate, followed by reductive elimination of Cu(I).[11] In the original paper describing this reaction, methylmagnesium bromide is reacted with isophorone with and without 1 mole percent of added copper(I) chloride (see figure).[10]
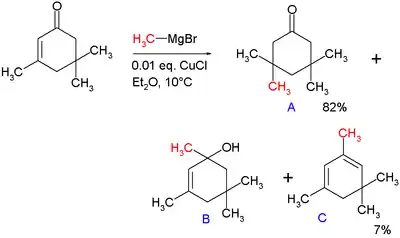
Without added salt the main products are alcohol B (42%) from nucleophilic addition to the carbonyl group and diene C (48%) as its dehydration reaction product. With added salt the main product is 1,4-adduct A (82%) with some C (7%).
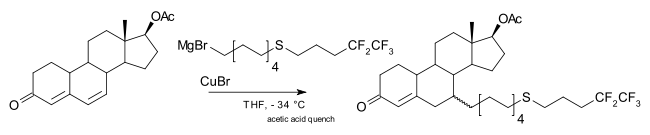
A 1,6-addition is also possible, for example in one step of the commercial-scale production of fulvestrant:[12]
Enantioselective Variants
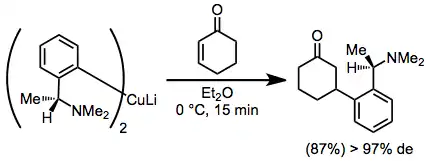
Diastereoselective conjugate addition reactions of chiral organocuprates provide β-functionalized ketones in high yield and diastereoselectivity. A disadvantage of these reactions is the requirement of a full equivalent of enantiopure starting material.[13]
(3)
More recently, catalytic enantioselective methods have been developed based on the copper(I)-catalyzed conjugate addition of Grignard reactions to enones. The proposed mechanism involves transmetalation from the Grignard reagent to copper, conjugate addition, and rate-determining reductive elimination (see the analogous upper pathway in equation (2)).[14]
(4)
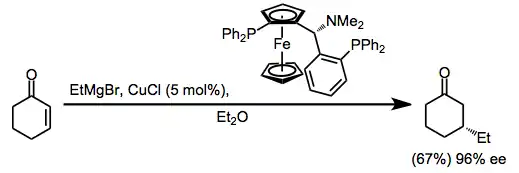
Catalytic reactions
Vinyl and aryl Grignard reagents couple with primary alkyl halides in the presence of a catalytic amount of a copper(I) halide salt. The use of Li2CuCl4 rather than simple copper(I) halide salts (CuX) improves yields of these coupling reactions.[15]
(5)
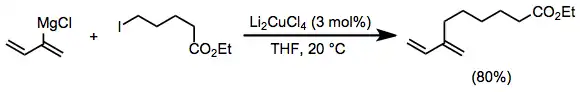
The addition of Grignard reagents to alkynes is facilitated by a catalytic amount of copper halide. Transmetalation to copper and carbocupration are followed by transmetalation of the product alkene back to magnesium. The addition is syn unless a coordinating group is nearby in the substrate, in which case the addition becomes anti and yields improve.[16]
(6)
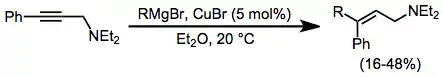
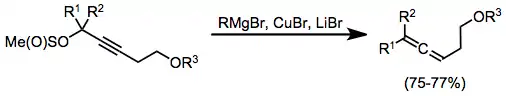
Stoichiometric reactions
Propargyl methanesulfinates are useful substrates for the synthesis of allenes from stoichiometric organocopper complexes. In this case, the complexes were generated in situ through the combination of a Grignard reagent, copper(I) bromide, and lithium bromide. Organocopper complexes very often need Lewis acid activation in order to react efficiently; magnesium bromide generated in situ serves as an activating Lewis acid in this case.[17]
(7)
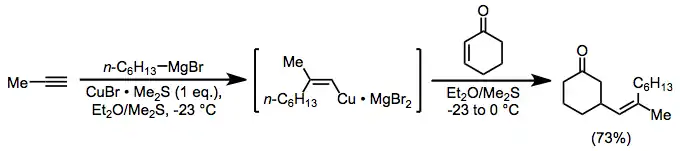
Alkenylcopper complexes, easily generated through carbocupration, are useful for the introduction of a vinyl group in the β position of a carbonyl compound. In this case, as above, magnesium bromide is serving as an activating Lewis acid.[18]
(8)
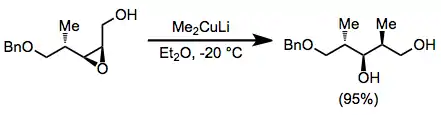
Epoxide opening with organocuprates is highly selective for the less hindered position. Substitution takes place with complete inversion of configuration at the electrophilic carbon.[19]
(9)
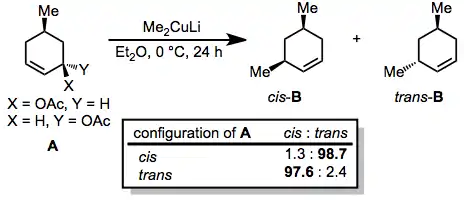
Generally, organocuprates react with allylic electrophiles in an anti SN2 fashion. In the reaction below, nearly complete inversion of configuration was observed despite the presence of a second stereocenter in the ring.[20]
(10)
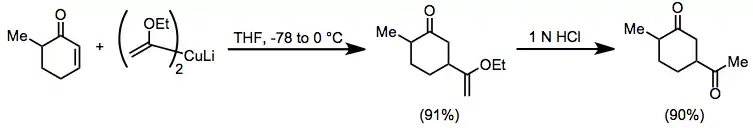
Conjugate addition of organocuprates is widely used in organic synthesis. Vinyl ether cuprates serve as convenient acyl anion equivalents in conjugate addition reactions to enones. The resulting enol ethers can be hydrolyzed to 1,4-diketones, which are difficult to access using conventional carbonyl chemistry.[21]
(11)
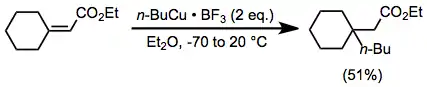
The use of additives in conjunction with a stoichiometric amount of organocopper complexes enhances the rate and yield of many reactions. Organocopper complexes in particular react sluggishly in the absence of a Lewis acid. Although magnesium bromide generated in situ from the reaction of Grignard reagents and copper(I) halides can serve this role (see above), external Lewis acids are also useful. In the presence of boron trifluoride etherate, organocopper complexes are able to add to sterically congested enones in moderate yield (effecting the same transformation with an organocuprate would be difficult).[22]
(12)
Boron trifluoride etherate is also useful as an additive in reactions of higher-order cyanocuprates. The use of the 2-thienyl group as a "dummy" substituent in the cyanocuprate conserves the potentially valuable organolithium reagent used to generate the cyanocuprate (as only the dummy group is present in copper-containing byproducts). In the absence of boron trifluoride etherate, no reaction was observed in this case.[23]
(13)
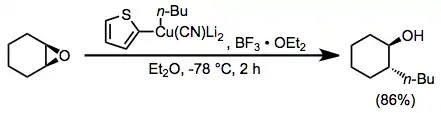
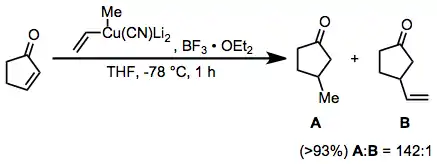
Conjugate addition reactions of higher-order cyanocuprates represent another useful application for boron trifluoride etherate. The vinyl group is transferred selectively in this reaction; this is in contrast to substitution reactions employing the same reagent, which result in selective transfer of the methyl group.[24]
(14)
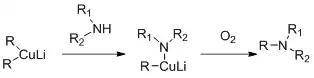
Alkylation of amines
Secondary amines can be alkylated with cuprates. The reaction is based on the oxidative coupling of lithium alkyl copper amide which is reported to form in situ during the reaction between lithium dialkylcuprates and primary or secondary amides.[25]
Synthetic Applications
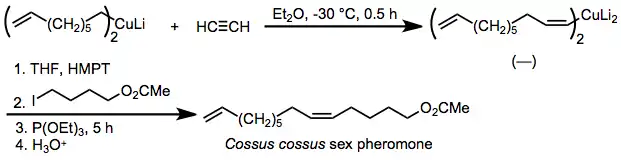
Because the stereoselectivity of carbocupration is extremely high, the reaction has been applied to the synthesis of pheromones in which the geometric purity of double bonds is critical. One example is the insect pheromone of Cossus cossus, which is synthesized by syn-selective carbocupration of acetylene and alkylation of the resulting organocuprate in the presence of added phosphite.[26]
(15)
References
- Lipshutz, B. H.; Sengupta, S. Org. React. 1992, 41, 135. doi:10.1002/0471264180.or041.02
- Kharasch, M. S.; Tawney, P. O. J. Am. Chem. Soc. 1941, 63, 2308.
- Kansal, V. K.; Taylor, R. J. K. J. Chem. Soc. Perkin Trans. 1 1984, 703.
- Posner, G. H. Org. React. 1975, 22, 253.
- Lipshutz, B. H.; Wilhelm, R. S.; Floyd, D. M. J. Am. Chem. Soc. 1981, 103, 7672.
- Tamura, M.; Kochi, J. K. J. Organomet. Chem. 1972, 42, 205.
- Corey, E. J.; Boaz, N. W. Tetrahedron Lett. 1984, 25, 3059.
- Johnson, C. R.; Dutra, G. A. J. Am. Chem. Soc. 1973, 95, 7777.
- Ashby, E. C.; Coleman, D. J. Org. Chem. 1987, 52, 4554.
- Kharasch, M. S.; Tawney, P. O. (1941). "Factors Determining the Course and Mechanisms of Grignard Reactions. II. The Effect of Metallic Compounds on the Reaction between Isophorone and Methylmagnesium Bromide". Journal of the American Chemical Society. 63 (9): 2308–2316. doi:10.1021/ja01854a005. ISSN 0002-7863.
- Nakamura, Eiichi; Mori, Seiji (2000). "Wherefore Art Thou Copper? Structures and Reaction Mechanisms of Organocuprate Clusters in Organic Chemistry". Angewandte Chemie. 39 (21): 3750–3771. doi:10.1002/1521-3773(20001103)39:21<3750::AID-ANIE3750>3.0.CO;2-L. PMID 11091452.
- Fulvestrant: From the Laboratory to Commercial-Scale Manufacture Eve J. Brazier, Philip J. Hogan, Chiu W. Leung, Anne O’Kearney-McMullan, Alison K. Norton, Lyn Powell, Graham E. Robinson, and Emyr G. Williams Organic Process Research & Development 2010, 14, 544–552 doi:10.1021/op900315j
- Malmberg, H.; Nilsson, M.; Ullenius, C. Tetrahedron Lett. 1982, 23, 3823.
- Harutyunyan, S.; López, F.; Browne, W.; Correa, A.; Peña, D.; Badorrey, R.; Meetsma, A.; Minnaard, A.; Feringa, B. L. J. Am. Chem. Soc. 2006, 128, 9103.
- Nunomoto, S.; Kawakami, Y.; Yamashita, Y. J. Org. Chem. 1983, 48, 1912.
- Jousseaume, B. Ph.D. Thesis, University of Bordeaux, France, 1977.
- Kleijn, H.; Elsevier, C. J.; Westmijze, H.; Meijer, J.; Vermeer, P. Tetrahedron Lett. 1979, 3101.
- Marfat, A.; McGuirk, P. R.; Helquist, P. J. Org. Chem. 1979, 44, 3888.
- Johnson, M. R.; Nakata, T.; Kishi, Y. Tetrahedron Lett. 1979, 4343.
- Goering, H. L.; Kantner, S. S. J. Org. Chem. 1981, 46, 2144.
- Boeckman, R. K.; Ramaiah, M. J. Org. Chem. 1977, 42, 1581.
- Yamamoto, Y.; Yamamoto, S.; Yatagai, S.; Ishihara, Y.; Maruyama, K. J. Org. Chem. 1982, 47, 119.
- Lipshutz, B. H.; Parker, D. A.; Kozlowski, J. A.; Nguyen, S. L. Tetrahedron Lett. 1984, 25, 5959.
- Lipshutz, B. H.; Wilhelm, R. S.; Kozlowski, J. A. J. Org. Chem. 1984, 49, 3938.
- Yamamoto, H.; Marouka, K. (1980). "Novel N-alkylation of amines with organocopper reagents". J. Org. Chem. 45: 2739–2740. doi:10.1021/jo01301a048.
- Cahiez, G.; Alexakis, A.; Normant, J. F. Tetrahedron Lett. 1978, 2027.