Nuclear ensemble approach
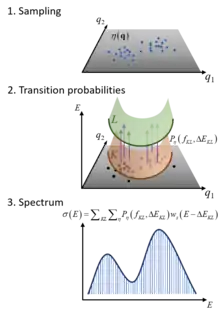
The Nuclear Ensemble Approach (NEA) is a general method for simulations of diverse types of molecular spectra.[1] It works by sampling an ensemble of molecular conformations (nuclear geometries) in the source state, computing the transition probabilities to the target states for each of these geometries, and performing a sum over all these transitions convoluted with shape function. The result is an incoherent spectrum containing absolute band shapes through inhomogeneous broadening.
Motivation
Spectrum simulation is one of the most fundamental tasks in quantum chemistry. It allows comparing the theoretical results to experimental measurements. There are many theoretical methods for simulating spectra. Some are simple approximations (like stick spectra); others are high-level, accurate approximations (like those based on Fourier-transform of wavepacket propagations). The NEA lies in between. On the one hand, it is intuitive and straightforward to apply, providing much improved results compared to the stick spectrum. On the other hand, it does not recover all spectral effects and delivers a limited spectral resolution.
Historical
The NEA is a multidimensional extension of the reflection principle,[2] an approach often used for estimating spectra in photodissociative systems. With popularization molecular mechanics, ensembles of geometries started to be also used to estimate the spectra through incoherent sums.[3] Thus, different from the reflection principle, which is usually done via direct integration of analytical functions, the NEA is a numerical approach. In 2012, a formal account of NEA showed that it corresponded to an approximation to the time-dependent spectrum simulation approach, employing a Monte Carlo integration of the wavepacket overlap time evolution.[1]
NEA for absorption spectrum
Consider an ensemble of molecules absorbing radiation in the UV/vis. Initially, all molecules are in the ground electronic state
Because of the molecular zero-point energy and temperature, the molecular geometry has a distribution around the equilibrium geometry. From a classical point of view, supposing that the photon absorption is an instantaneous process, each time a molecule is excited, it des from a different geometry. As a consequence, the transition energy has not always the same value, but is a function of the nuclear coordinates.
The NEA captures this effect by creating an ensemble of geometries reflecting the zero-point energy, the temperature, or both.
In the NEA, the absorption spectrum (or absorption cross section) σ(E) at excitation energy E is calculated as[1]

where e and m are the electron charge and mass, c is the speed of light, ε0 the vacuum permittivity, and ћ the reduced Planck constant. The sums run over Nfs excited states and Np nuclear geometries xi. For each of such geometries in the ensemble, transition energies ΔE0n(xi) and oscillator strengths f0n(xi) between the ground (0) and the excited (n) states are computed. Each transition in the ensemble is convoluted with a normalized line shape function centered at ΔE0n(xi) and with width δ. Each xi is a vector collecting the cartesian components of the geometries of each atom.
The line shape function may be, for instance, a normalized Gaussian function given by

Although δ is an arbitrary parameter, it must be much narrower than the band width, not to interfere in its description. As the average value of band widths is around 0.3 eV, it is a good practice to adopt δ ≤ 0.05 eV.[4]
The geometries xi can be generated by any method able to describe the ground state distribution. Two of the most employed are dynamics and Wigner distribution nuclear normal modes.[5]
Molar extinction coefficient ε can be obtained from absorption cross section through

Because of the dependence of f0n on xi, NEA is a post-Condon approximation, and it can predict dark vibronic bands.[1]
NEA for emission spectrum
In the case of fluorescence, the differential emission rate is given by[1]
.

This expression assumes the validity of the Kasha's rule, with emission from the first excited state.
NEA for other types of spectrum
NEA can be used for many types of steady-state and time-resolved spectrum simulations.[6] Some examples beyond absorption and emission spectra are:
Limitations of NEA
By construction, NEA does not include information about the target (final) states. For this reason, any spectral information that depends on these states cannot be described in the framework of NEA. For example, vibronically resolved peaks in the absorption spectrum will not appear in the simulations, only the band envelope around them, because these peaks depend on the wavefunction overlap between the ground and excited state.[11] NEA can be, however, coupled to excited-state dynamics to recover these effects.[12]
NEA may be too computationally expensive for large molecules. The spectrum simulation requires the calculation of transition probabilities for hundreds of different nuclear geometries, which may become prohibitive due to the high computational costs. Machine learning methods coupled to NEA have been proposed to reduce these costs.[13][4]
References
- Crespo-Otero R, Barbatti M (June 2012). "Spectrum simulation and decomposition with nuclear ensemble: formal derivation and application to benzene, furan and 2-phenylfuran". Theoretical Chemistry Accounts. 131 (6): 1237. doi:10.1007/s00214-012-1237-4. ISSN 1432-881X. S2CID 95758853.
- Schinke R (April 1993). Photodissociation Dynamics: Spectroscopy and Fragmentation of Small Polyatomic Molecules (1 ed.). Cambridge University Press. doi:10.1017/cbo9780511586453. ISBN 978-0-521-48414-5.
- Bergsma JP, Berens PH, Wilson KR, Fredkin DR, Heller EJ (February 1984). "Electronic spectra from molecular dynamics: a simple approach". The Journal of Physical Chemistry. 88 (3): 612–619. doi:10.1021/j150647a05 (inactive 2021-01-15).CS1 maint: DOI inactive as of January 2021 (link)
- Xue BX, Barbatti M, Dral PO (August 2020). "Machine Learning for Absorption Cross Sections". The Journal of Physical Chemistry A. 124 (35): 7199–7210. Bibcode:2020JPCA..124.7199X. doi:10.1021/acs.jpca.0c05310. PMC 7511037. PMID 32786977.
- Barbatti M, Sen K (May 2016). "Effects of different initial condition samplings on photodynamics and spectrum of pyrrole" (PDF). International Journal of Quantum Chemistry. 116 (10): 762–771. doi:10.1002/qua.25049.
- Bennett K, Kowalewski M, Mukamel S (2015). "Probing electronic and vibrational dynamics in molecules by time-resolved photoelectron, Auger-electron, and X-ray photon scattering spectroscopy". Faraday Discussions. 177: 405–28. Bibcode:2015FaDi..177..405B. doi:10.1039/C4FD00178H. PMC 4401660. PMID 25730500.
- Segarra-Martí J, Segatta F, Mackenzie TA, Nenov A, Rivalta I, Bearpark MJ, Garavelli M (December 2019). "Modeling multidimensional spectral lineshapes from first principles: application to water-solvated adenine". Faraday Discussions. 221: 219–244. doi:10.1039/C9FD00072K. hdl:10044/1/70922. PMID 31544178.
- Polli D, Altoè P, Weingart O, Spillane KM, Manzoni C, Brida D, et al. (September 2010). "Conical intersection dynamics of the primary photoisomerization event in vision". Nature. 467 (7314): 440–3. Bibcode:2010Natur.467..440P. doi:10.1038/nature09346. PMID 20864998. S2CID 4354278.
- Arbelo-González W, Crespo-Otero R, Barbatti M (October 2016). "Steady and Time-Resolved Photoelectron Spectra Based on Nuclear Ensembles" (PDF). Journal of Chemical Theory and Computation. 12 (10): 5037–5049. doi:10.1021/acs.jctc.6b00704. PMID 27588827.
- McFarland BK, Farrell JP, Miyabe S, Tarantelli F, Aguilar A, Berrah N, et al. (June 2014). "Ultrafast X-ray Auger probing of photoexcited molecular dynamics". Nature Communications. 5 (1): 4235. Bibcode:2014NatCo...5.4235M. doi:10.1038/ncomms5235. PMID 24953740.
- Heller EJ (December 1981). "The semiclassical way to molecular spectroscopy 2". Accounts of Chemical Research. 14 (12): 368–375. doi:10.1021/ar00072a002.
- Petit AS, Subotnik JE (October 2014). "Calculating time-resolved differential absorbance spectra for ultrafast pump-probe experiments with surface hopping trajectories". The Journal of Chemical Physics. 141 (15): 154108. Bibcode:2014JChPh.141o4108P. doi:10.1063/1.4897258. PMID 25338882.
- Ye ZR, Huang IS, Chan YT, Li ZJ, Liao CC, Tsai HR, et al. (2020). "Predicting the emission wavelength of organic molecules using a combinatorial QSAR and machine learning approach". RSC Advances. 10 (40): 23834–23841. Bibcode:2020RSCAd..1023834Y. doi:10.1039/D0RA05014H.