Nitroxide-mediated radical polymerization
Nitroxide-mediated radical polymerization is a method of radical polymerization that makes use of an nitroxide initiator to generate polymers with well controlled stereochemistry and a very low dispersity.[1] It is a type of reversible-deactivation radical polymerization.
Alkoxyamine Initiators
The initiating materials for nitroxide-mediated radical polymerization (NMP) are a family of compounds referred to as alkoxyamines. An alkoxyamine can essentially be viewed as an alcohol bound to a secondary amine by an N-O single bond. The utility of this functional group is that under certain conditions, homolysis of the C-O bond can occur, yielding a stable radical in the form of a 2-center 3-electron N-O system and a carbon radical which serves as an initiator for radical polymerization.[2] For the purposes of NMP, the R groups attached to the nitrogen are always bulky, sterically hindering groups and the R group in the O- position forms a stable radical, generally is benzylic for polymerization to occur successfully. NMP allows for excellent control of chain length and structure, as well as a relative lack of true termination that allows polymerization to continue as long as there is available monomer. Because of this it is said to be “living".
Persistent radical effect
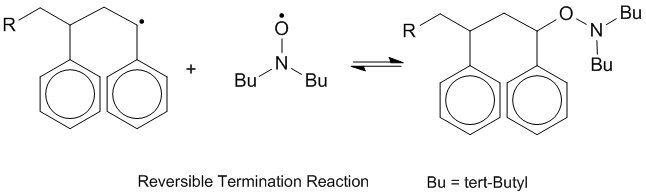
The living nature of NMP is due to the persistent radical effect (PRE).[3] The PRE is a phenomenon observable in some radical systems which leads to the highly favored formation of one product to the near exclusion of other radical couplings due to one of the radical species being particularly stable, existing in greater and greater concentrations as the reaction progresses while the other one is transient, reacting quickly with either itself in a termination step or with the persistent radical to form a desired product. As time goes on, a higher concentration of the persistent radical is present, which couples reversibly with itself, meaning that any of the transient radical still present tends to couple with the persistent radical rather than itself due to greater availability. This leads to a greater proportion of cross-coupling than self-coupling in radical species.[4]
In the case of a nitroxide-mediated polymerization reaction, the persistent radical is the nitroxide species and the transient radical is always the carbon radical. This leads to repeated coupling of the nitroxide to the growing end of the polymer chain, which would ordinarily be considered a termination step, but is in this case reversible. Because of the high rate of coupling of the nitroxide to the growing chain end, there is little coupling of two active growing chains, which would be an irreversible terminating step limiting the chain length. The nitroxide binds and unbinds to the growing chain, protecting it from termination steps. This ensures that any available monomer can be easily scavenged by active chains. Because this polymerization process does not naturally self-terminate, this polymerization process is described as “living,” as the chains continue to grow under suitable reaction conditions whenever there is reactive monomer to “feed” them. Because of the PRE, it can be assumed that at any given time, almost all of the growing chains are “capped” by a mediating nitroxide, meaning that they dissociate and grow at very similar rates, creating a largely uniform chain length and structure.[5]
Nitroxide stability
As stated above, nitroxide radicals are effective mediators of well-controlled radical polymerization because they are quite stable, allowing them to act as persistent radicals in a reaction mixture. This stability is a result of their unique structure. In most diagrams, the radical is depicted on the oxygen, but another resonance structure exists which is more helpful in explaining their stability in which the radical is on the nitrogen, which has a double bond to the oxygen. In addition to this resonance stability, nitroxides used in NMRP always contain bulky, sterically hindering groups in the R1 and R2 positions. The significant steric bulk of these substituents entirely prevents radical coupling in the N-centered resonance form while significantly reducing it in the O-centered form. These bulky groups contribute stability, but only if there is no resonance provided by allyl or aromatic groups α to the N. These result in decreased stability of the nitroxide, presumably because they offer less sterically hindered sites for radical coupling to take place.[6] The resulting inactivity of the radical makes hemolytic cleavage of the alkoxyamine quite fast in more sterically hindered species.[7]
Nitroxide choice
The choice of a specific nitroxide species to use has a large effect on the efficacy of an attempted polymerization. An effective polymerization (fast rate of chain growth, consistent chain length) results from a nitroxide with a fast C-O homolysis and relatively few side reactions. A more polar solvent lends itself better to C-O homolysis, so polar solvents which cannot bind to a labile nitroxide are the most effective for NMP. It is generally agreed that the structural factor that has the greatest effect on the ability of a nitroxide to mediate a radical polymerization is steric bulk. Generally speaking, greater steric bulk on the nitroxide leads to greater strain on the alkoxyamine, leading to the most easily broken bond, the C-O single bond, cleaving homolytically.
Ring size
In the case of cyclic nitroxides, five-membered ring systems have been shown to cleave more slowly than six-membered rings and acyclic nitroxides with t-butyl moieties as their R groups cleaved fastest of all. This difference in the rate of cleavage was determined to result not from a difference in C-O bond lengths, but in the difference of C-O-N bond angle in the alkoxyamine. The smaller the bond angle the greater the steric interaction between the nitroxide and the alkyl fragment and the more easily the initiator species broke apart.[8]
Steric bulk
The efficiency of polymerization increases more and more with increased steric bulk of the nitroxide up to a point. TEMPO ((2,2,6,6-Tetramethylpiperidin-1-yl)oxyl) is capable of inducing the polymerization of styrene and styrene derivatives fairly easily, but is not sufficiently labile to induce polymerization of butyl acrylate under most conditions. TEMPO derivatives with even bulkier groups at the positions α to N have a rate of homolysis great enough to induce NMP of butyl acrylate, and the bulkier the α groups, the faster polymerization occurs. This indicates that the steric bulk of the nitroxide fragment can be a good indicator of the strength of an alkoxyamine initiator, at least up to a point. The equilibrium of its homolysis and reformation favors the radical form to the extent that recombination to reform an alkoxyamine over the course of NMP occurs too slowly to maintain control of chain length.[9]
Preparation methods
Because TEMPO, which is commercially available, is a sufficient nitroxide mediator for the synthesis of polystyrene derivatives, the preparation of alkoxyamine initiators for NMP of copolymers is in many cases a matter of attaching a nitroxide group (TEMPO) to a specifically synthesized alkyl fragment. Several methods have been reported to achieve this transformation.
Jacobsen's catalyst
Jacobsen's catalyst is a manganese-based catalyst commonly used for the stereoselective epoxidation of alkenes. This epoxidation proceeds by a radical addition mechanism, which can be taken advantage of by introducing the radical TEMPO group into the reaction mixture. After treatment with a mild reducing agent such as sodium borohydride, this yields the product of a Markovnikov addition of nitroxide to the alkene. Jacobsen’s catalyst is fairly mild, and a wide variety of functionalities on the alkene substrate can be tolerated. Practical yields are not necessarily as high as those reported by Dao et al., however.[10]
Hydrazine
An alternative method is to react a substrate with a C-Br bond at the desired location of the nitroxide with hydrazine, generating an alkyl substituted hydrazine which is then exposed to a nitroxide radical and a mild oxidating agent such as lead dioxide. This generates a carbon-centered radical which couples with the nitroxide to generate the desired alkoxyamine. This method has the disadvantage of being relatively inefficient for some species, as well as the inherent danger of having to work with extremely toxic hydrazine and the inconvenience of having to run reactions in inert atmosphere.[11]
Treatment of aldehydes with hydrogen peroxide
Yet another published alkoxyamine synthesis involves treatment of aldehydes with hydrogen peroxide, which adds to the carbonyl group. The resulting species rearranges in situ in the presence of CuCl forming formic acid and the desired alkyl radical, which couples with tempo to produce the target alkoxyamine. The reaction appears to give fairly good yields and tolerates a variety of functional groups in the alkyl chain.[12]
Electrophilic bromination and nucleophilic attack
A synthesis has been described by Moon and Kang consisting of a one-electron reduction of a nitroxide radical in metallic sodium to yield a nucleophilic nitroxide. The nitroxide nucleophile is then added to an appropriate alkyl bromide, yielding the alkoxyamine by a simple SN2 reaction. This technique has the advantage of requiring only the appropriate alkyl bromide to be synthesized without requiring inconvenient reaction conditions and extremely hazardous reagents like Braso et al.’s method.[13]
References
- Nicolas, J., et al. Prog. Polym. Sci., 2013, 38, 63–235
- Moad, G., Rizzardo, E. Macromolecules, 1995, 28, 8722–8728.
- Bertin, D., et al. Chem. Soc. Rev., 2011, 40, 2189–2198
- Fischer, Hanns. Chem. Rev., 2001, 101 (12), 3581–3610.
- Hawker, C.J., Barclay, G.G., Dao, J.J. Am. Chem. Soc., 1996, 118 (46), 11467–11471.
- Volodarsky, L.B., Reznikov, V.A., Ovcharenko, V.I. Synthetic Chemistry of Stable Nitroxides. CRC Press, 1994.
- Bertin, D., et al. Chem. Soc. Rev., 2011, 40, 2189–2198
- Moad, G., Rizzardo, E. Macromolecules, 1995, 28, 8722–8728.
- Siegenthaler, K.O., Studer, A. Macromolecules, 2006, 39(4), 1347–1352.
- Dao, J., Benoit, D., Hawker, C.J.J. Poly. Sci., 1998, 36, 2161–2167.
- Braslo, R., et al. Macromolecules, 1997, 30, 6445–6450.
- Schoening, K.U., et al. J. Org. Chem. 2009, 74, 1567–1573.
- Moon, B., Minjyuk, K. Macromol. Res., 2005, 13(3), 229–235.