Molecular ecology
Molecular ecology is a field of evolutionary biology[1] that is concerned with applying molecular population genetics, molecular phylogenetics, and more recently genomics to traditional ecological questions (e.g., species diagnosis, conservation and assessment of biodiversity, species-area relationships, and many questions in behavioral ecology). It is virtually synonymous with the field of "Ecological Genetics" as pioneered by Theodosius Dobzhansky, E. B. Ford, Godfrey M. Hewitt, and others.[2] These fields are united in their attempt to study genetic-based questions "out in the field" as opposed to the laboratory. Molecular ecology is related to the field of conservation genetics.
Methods frequently include using microsatellites to determine gene flow and hybridization between populations. The development of molecular ecology is also closely related to the use of DNA microarrays, which allows for the simultaneous analysis of the expression of thousands of different genes. Quantitative PCR may also be used to analyze gene expression as a result of changes in environmental conditions or different responses by differently adapted individuals.
Molecular ecology uses molecular genetic data to answer ecological question related to biogeography, genomics, conservation genetics, and behavioral ecology. Studies mostly use data based on deoxyribonucleic acid sequences (DNA). This approach has been enhanced over a number of years to allow researchers to sequence thousands of genes from a small amount of starting DNA. Allele sizes are another way researchers are able to compare individuals and populations which allows them to quantify the genetic diversity within a population and the genetic similarities among populations.[3]

Bacterial diversity
Molecular ecological techniques are used to study in situ questions of bacterial diversity. Many microorganisms are not easily obtainable as cultured strains in the laboratory, which would allow for identification and characterization. It also stems from the development of PCR technique, which allows for the rapid amplification of genetic material.
The amplification of DNA from environmental samples using general or group-specific primers leads to a mix of genetic material, requiring sorting before sequencing and identification. The classic technique to achieve this is through cloning, which involves incorporating the amplified DNA fragments into bacterial plasmids. Techniques such as temperature gradient gel electrophoresis, allow for a faster result. More recently, the advent of relatively low-cost, next-generation DNA sequencing technologies, such as 454 and Illumina platforms, has allowed exploration of bacterial ecology concerning continental-scale environmental gradients such as pH[4] that was not feasible with traditional technology.

Fungal diversity
Exploration of fungal diversity in situ has also benefited from next-generation DNA sequencing technologies. The use of high-throughput sequencing techniques has been widely adopted by the fungal ecology community since the first publication of their use in the field in 2009.[5] Similar to the exploration of bacterial diversity, these techniques have allowed high-resolution studies of fundamental questions in fungal ecology such as phylogeography,[6] fungal diversity in forest soils,[7] stratification of fungal communities in soil horizons,[8] and fungal succession on decomposing plant litter.[9]
The majority of fungal ecology research leveraging next-generation sequencing approaches involves sequencing of PCR amplicons of conserved regions of DNA (i.e. marker genes) to identify and describe the distribution of taxonomic groups in the fungal community in question, though more recent research has focused on sequencing functional gene amplicons[5] (e.g. Baldrian et al. 2012[8]). The locus of choice for a description of the taxonomic structure of fungal communities has traditionally been the internal transcribed spacer (ITS) region of ribosomal RNA genes [10] due to its utility in identifying fungi to genus or species taxonomic levels,[11] and its high representation in public sequence databases.[10] A second widely used locus (e.g. Amend et al. 2010,[6] Weber et al. 2013[12]), the D1-D3 region of 28S ribosomal RNA genes, may not allow the low taxonomic level classification of the ITS,[13][14] but demonstrates superior performance in sequence alignment and phylogenetics.[6][15] Also, the D1-D3 region may be a better candidate for sequencing with Illumina sequencing technologies.[16] Porras-Alfaro et al.[14] showed that the accuracy of classification of either ITS or D1-D3 region sequences was largely based on the sequence composition and quality of databases used for comparison, and poor-quality sequences and sequence misidentification in public databases is a major concern.[17][18] The construction of sequence databases that have broad representation across fungi, and that are curated by taxonomic experts is a critical next step.[15][19]
Next-generation sequencing technologies generate large amounts of data, and analysis of fungal marker-gene data is an active area of research.[5][20] Two primary areas of concern are methods for clustering sequences into operational taxonomic units by sequence similarity and quality control of sequence data.[5][20] Currently, there is no consensus on preferred methods for clustering,[20] and clustering and sequence processing methods can significantly affect results, especially for the variable-length ITS region.[5][20] In addition, fungal species vary in intra-specific sequence similarity of the ITS region.[21] Recent research has been devoted to the development of flexible clustering protocols that allow sequence similarity thresholds to vary by taxonomic groups, which are supported by well-annotated sequences in public sequence databases.[19]
Extra-pair fertilizations
In recent years, molecular data and analyses have been able to supplement traditional approaches of behavioral ecology, the study of animal behavior in relation to its ecology and evolutionary history. One behavior that molecular data has helped scientists better understand is extra-pair fertilizations (EPFs), also known as extra-pair copulations (EPCs). These are mating events that occur outside of a social bond, like monogamy and are hard to observe. Molecular data has been key to understanding the prevalence of and the individuals participating in EPFs.
While most bird species are socially monogamous, molecular data has revealed that less than 25% of these species are genetically monogamous.[22] EPFs complicate matters, especially for male individuals, because it does not make sense for an individual to care for offspring that are not their own. Studies have found that males will adjust their parental care in response to changes in their paternity.[23][24] Other studies have shown that in socially monogamous species, some individuals will employ an alternative strategy to be reproductively successful since a social bond does not always equal reproductive success.[25][26]
It appears that EPFs in some species is driven by the good genes hypothesis,[27]:295 In red-back shrikes (Lanius collurio) extra-pair males had significantly longer tarsi than within-pair males, and all of the extra-pair offspring were males, supporting the prediction that females will bias their clutch towards males when they mate with an "attractive" male.[28] In house wrens (Troglodytes aedon), extra-pair offspring were also found to be male-biased compared to within-offspring.[29]
Without molecular ecology, identifying individuals that participate in EPFs and the offspring that result from EPFs would be impossible.
Isolation by distance
Isolation by distance (IBD), like reproductive isolation, is the effect of physical barriers to populations that limit migration and lower gene flow. The shorter the distance between populations the more likely individuals are to disperse and mate and thus, increase gene flow.[30] The use of molecular data, specifically allele frequencies of individuals among populations in relation to their geographic distance help to explain concepts such as, sex-biased dispersal, speciation, and landscape genetics.
The Mantel test is an assessment that compares genetic distance with geographic distance and is most appropriate because it doesn't assume that the comparisons are independent of each other.[3]:135 There are three main factors that influence the chances of finding a correlation of IBD, which include sample size, metabolism, and taxa.[31] For example, based on the meta-analysis, ectotherms are more likely than endotherms to display greater IBD.
Metapopulation theory
Metapopulation theory dictates that a metapopulation consists of spatially distinct populations that interact with one another on some level and move through a cycle of extinctions and recolonizations (i.e. through dispersal).[32] The most common metapopulation model is the extinction-recolonization model which explains systems in which spatially distinct populations undergo stochastic changes in population sizes which may lead to extinction at the population level. Once this has occurred, dispersing individuals from other populations will immigrate and "rescue" the population at that site. Other metapopulation models include the source-sink model (island-mainland model) where one (or multiple) large central population(s) produces disperses to smaller satellite populations that have a population growth rate of less than one and could not persist without the influx from the main population.
Metapopulation structure and the repeated extinctions and recolonizations can significantly affect a population's genetic structure. Recolonization by a few dispersers leads to population bottlenecks which will reduce the effective population size (Ne), accelerate genetic drift, and deplete genetic variation. However, dispersal between populations in the metapopulation can reverse or halt these processes over the long term. Therefore, in order for individual sub-populations to remain healthy, they must either have a large population size or have a relatively high rate of dispersal with other subpopulations. Molecular ecology focuses on using tests to determine the rates of dispersal between populations and can use molecular clocks to determine when historic bottlenecks occurred. As habitat becomes more fragmented, dispersal between populations will become increasingly rare. Therefore, subpopulations that may have historically been preserved by a metapopulation structure may start to decline. Using mitochondrial or nuclear markers to monitor dispersal coupled with population Fst values and allelic richness can provide insight into how well a population is performing and how it will perform into the future.
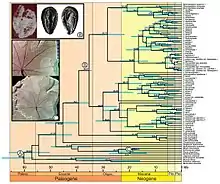
Molecular clock hypothesis
The molecular clock hypothesis states that DNA sequences roughly evolve at the same rate and because of this the dissimilarity between two sequences can be used to tell how long ago they diverged from one another. The first step in using a molecular clock is it must be calibrated based on the approximate time the two lineages studied diverged. The sources usually used to calibrate the molecular clocks are fossils or known geological events in the past. After calibrating the clock the next step is to calculate divergence time by dividing the estimated time since the sequences diverged by the amount of sequence divergence. The resulting number is the estimated rate at which molecular evolution is occurring. The most widely cited molecular clock is a ‘universal’ mtDNA clock of approximately two percent sequence divergence every million years.[33] Although referred to as a universal clock, this idea of the "universal" clock is not possible considering rates of evolution differ within DNA regions. Another drawback to using molecular clocks is that they ideally need to be calibrated from an independent source of data other than the molecular data. This poses a problem for taxa that don't fossilize/preserve easily, making it almost impossible to calibrate their molecular clock. Despite these inconveniences, the molecular clock hypothesis is still used today. The molecular clock has been successful in dating events happening up to 65 million years ago.[34]
%252C_mating.jpg.webp)
Mate choice hypotheses
The concept of mate choice explains how organisms select their mates based on two main methods; The Good Genes Hypothesis and Genetic Compatibility. The Good Genes Hypothesis, also referred to as the sexy son hypothesis, suggests that the females will choose a male that produce an offspring that will have increased fitness advantages and genetic viability. Therefore, the mates that are more 'attractive" are more likely to be chosen for mating and pass on their genes to the next generation. In species which exhibit polyandry the females will search out for the most suitable males and re-mate until they have found the best sperm to fertilize their eggs.[35] Genetic compatibility is where mates are choosing their partner based on the compatibility of their genotypes. The mate which is doing the selecting must know their own genotype as well as the genotypes of potential mates in order to select the appropriate partner.[36] Genetic compatibility in most instances is limited to specific traits, such as the major histocompatibility complex in mammals, because of complex genetic interactions. This behavior is potentially seen in humans. A study looking at women's choice in men based on body odors concluded that the scent of the odors were influenced by the MHC and that they influence mate choice in human populations.[37]
Sex-biased dispersal
Sex-biased dispersal, or the tendency of one sex to disperse between populations more frequently than the other, is a common behavior studied by researchers. Three major hypotheses currently exist to help explain sex-biased dispersal.[38] The resource-competition hypothesis infers that the more philopatric sex (the sex more likely to remain at its natal grounds) benefits during reproduction simply by having familiarity with natal ground resources.[39] A second proposal for sex-biased dispersal is the local mate competition hypothesis, which introduces the idea that individuals encounter less mate competition with relatives the farther from their natal grounds they disperse.[40] And the inbreeding avoidance hypothesis suggests individuals disperse to decrease inbreeding.
Studying these hypotheses can be arduous since it is nearly impossible to keep track of every individual and their whereabouts within and between populations. To combat this time-consuming method, scientists have recruited several molecular ecology techniques in order to study sex-biased dispersal. One method is the comparison of differences between nuclear and mitochondrial markers among populations. Markers showing higher levels of differentiation indicate the more philopatric sex; that is, the more a sex remains at natal grounds, the more their markers will take on a unique I.D, due to lack of gene flow with respect to that marker.[41] Researchers can also quantify male-male and female-female pair relatedness within populations to understand which sex is more likely to disperse. Pairs with values consistently lower in one sex indicate the dispersing sex. This is because there is more gene flow in the dispersing sex and their markers are less similar than individuals of the same sex in the same population, which produces a low relatedness value.[42] FST values are also used to understand dispersing behaviors by calculating an FST value for each sex. The sex that disperses more displays a lower FST value, which measures levels of inbreeding between the subpopulation and the total population. Additionally, assignment tests can be utilized to quantify the number of individuals of a certain sex dispersing to other populations. A more mathematical approach to quantifying sex-biased dispersal on the molecular level is the use of spatial autocorrelation.[43] This correlation analyzes the relationship between geographic distance and spatial distance. A correlation coefficient, or r value, is calculated and the plot of r against distance provides an indication of individuals more related to or less related to one another than expected.[27]:299–307
Quantitative trait loci
A quantitative trait locus (QTL) refers to a suite of genes that controls a quantitative trait. A quantitative trait is one that is influenced by several different genes as opposed to just one or two.[27] QTLs are analyzed using Qst. Qst looks at the relatedness of the traits in focus. In the case of QTLs, clines are analyzed by Qst. A cline (biology) is a change in allele frequency across a geographical distance.[27] This change in allele frequency causes a series of intermediate varying phenotypes that when associated with certain environmental conditions can indicate selection. This selection causes local adaptation, but high gene flow is still expected to be present along the cline.
For example, barn owls in Europe exhibit a cline in reference to their plumage coloration. Their feathers range in coloration from white to reddish-brown across the geological range of the southwest to the northeast.[44] This study sought to find if this phenotypic variation was due to selection by calculating the Qst values across the owl populations. Because high gene flow was still anticipated along this cline, selection was only expected to act upon the QTLs that incur locally adaptive phenotypic traits. This can be determined by comparing the Qst values to Fst (fixation index) values. If both of these values are similar and Fst is based on neutral markers then it can be assumed that the QTLs were based on neutral markers (markers not under selection or locally adapted) as well. However, in the case of the barn owls the Qst value was much higher than the Fst value. This means that high gene flow was present allowing the neutral markers to be similar, indicated by the low Fst value. But, local adaptation due to selection was present as well, in the form of varying plumage coloration since the Qst value was high, indicating differences in these non-neutral loci.[44] In other words, this cline of plumage coloration has some sort of adaptive value to the birds.
Fixation indices
Fixation indices are used when determining the level of genetic differentiation between sub-populations within a total population. FST is the script used to represent this index when using the formula:

In this equation, HT represents the expected heterozygosity of the total population and HS is the expected heterozygosity of a sub-populations. Both measures of heterozygosity are measured at one loci. In the equation, heterozygosity values expected from the total population are compared to observed heterozygosity values of the sub-populations within this total population. Larger FST values imply that the level of genetic differentiation between sub-populations within a total population is more significant.[27] The level of differentiation is the result of a balance between gene flow amongst sub-populations (decreasing differentiation) and genetic drift within these sub-populations (increasing differentiation); however, some molecular ecologists note that it cannot be assumed that these factors are at equilibrium.[45] FST can also be viewed as a way of comparing the amount of inbreeding within sub-populations to the amount of inbreeding for the total population and is sometimes referred to as an inbreeding coefficient. In these cases, higher FST values typically imply higher amounts of inbreeding within the sub-populations.[46] Other factors such as selection pressures may also affect FST values.[47]
FST values are accompanied by several analog equations (FIS, GST, etc.). These additional measures are interpreted in a similar manner to FST values; however, they are adjusted to accompany other factors that FST may not, such as accounting for multiple loci.[48]
_(14579196860).jpg.webp)
Inbreeding depression
Inbreeding depression is the reduced fitness and survival of offspring from closely related parents.[49] Inbreeding is commonly seen in small populations because of the greater chance of mating with a relative due to limited mate choice. Inbreeding, especially in small populations, is more likely to result in higher rates of genetic drift, which leads to higher rates of homozygosity at all loci in the population and decreased heterozygosity. The rate of inbreeding is based on decreased heterozygosity. In other words, the rate at which heterozygosity is lost from a population due to genetic drift is equal to the rate of accumulating inbreeding in a population. In the absence of migration, inbreeding will accumulate at a rate that is inversely proportional to the size of the population.
There are two ways in which inbreeding depression can occur. The first of these is through dominance, where beneficial alleles are usually dominant and harmful alleles are usually recessive. The increased homozygosity resulting from inbreeding means that harmful alleles are more likely to be expressed as homozygotes, and the deleterious effects cannot be masked by the beneficial dominant allele. The second method through which inbreeding depression occurs is through overdominance, or heterozygote advantage.[50] Individuals that are heterozygous at a particular locus have a higher fitness than homozygotes at that locus. Inbreeding leads to decreased heterozygosity, and therefore decreased fitness.
Deleterious alleles can be scrubbed by natural selection from inbred populations through genetic purging. As homozygosity increases, less fit individuals will be selected against and thus those harmful alleles will be lost from the population.[27]
Outbreeding depression
Outbreeding depression is the reduced biological fitness in the offspring of distantly related parents.[51] The decline in fitness due to outbreeding is attributed to a breakup of coadapted gene complexes or favorable epistatic relationships. Unlike inbreeding depression, outbreeding depression emphasizes interactions between loci rather than within them. Inbreeding and outbreeding depression can occur at the same time. Risks of outbreeding depression increase with increased distance between populations. The risk of outbreeding depression during genetic rescue often limits the ability to increase a small or fragmented gene pool's genetic diversity.[51] The spawn of an intermediate of two or more adapted traits can render the adaptation less effective than either of the parental adaptations.[52] Three main mechanisms influence outbreeding depression; genetic drift, population bottlenecking, differentiation of adaptations, and set chromosomal dissimilarities resulting in sterile offspring.[53] If outbreeding is limited and the population is large enough, selective pressure acting on each generation may be able to restore fitness. However, the population is likely to experience a multi-generational decline of overall fitness as selection for traits takes multiple generations.[54] Selection acts on outbred generations using increased diversity to adapt to the environment. This may result in greater fitness among offspring than the original parental type.
Conservation units
Conservation units are classifications often used in conservation biology, conservation genetics, and molecular ecology in order to separate and group different species or populations based on genetic variance and significance for protection.[55] Two of the most common types of conservation units are:
- Management Units (MU): Management units are populations that have very low levels of gene flow and can therefore be genetically differentiated from other populations.[55]
- Evolutionarily significant units (ESU): Evolutionarily significant units are populations that show enough genetic differentiation to warrant their management as distinct units.[55]
Conservation units are often identified using both neutral and non-neutral genetic markers, with each having its own advantages. Using neutral markers during unit identification can provide unbiased assumptions of genetic drift and time since reproductive isolation within and among species and populations, while using non-neutral markers can provide more accurate estimations of adaptive evolutionary divergence, which can help determine the potential for a conservation unit to adapt within a certain habitat.[55]
Because of conservation units, populations and species that have high or differing levels of genetic variation can be distinguished in order to manage each individually, which can ultimately differ based on a number of factors. In one instance, Atlantic salmon located within the Bay of Fundy were given evolutionary significance based on the differences in genetic sequences found among different populations.[56] This detection of evolutionary significance can allow each population of salmon to receive customized conservation and protection based on their adaptive uniqueness in response to geographic location.[56]

Phylogenies and community Ecology
Phylogenies are the evolutionary history of an organism, also known as phylogeography. A phylogenetic tree is a tree that shows evolutionary relationships between different species based on similarities/differences among genetic or physical traits. Community ecology is based on knowledge of evolutionary relationships among coexisting species.[57] Phylogenies embrace aspects of both time (evolutionary relationships) and space (geographic distribution).[58] Typically phylogeny trees include tips, which represent groups of descendent species, and nodes, which represent the common ancestors of those descendants. If two descendants split from the same node, they are called sister groups. They also may include an outgroup, a species outside of the group of interest.[59] The trees depict clades, which is a group of organisms that include an ancestor and all descendants of that ancestor. The maximum parsimony tree is the simplest tree that has the minimum number of steps possible.
Phylogenies confer important historical processes that shape current distributions of genes and species.[58] When two species become isolated from each other they retain some of the same ancestral alleles also known as allele sharing. Alleles can be shared because of lineage sorting and hybridization. Lineage sorting is driven by genetic drift and must occur before alleles become species specific. Some of these alleles over time will simply be lost, or they may proliferate. Hybridization leads to introgression of alleles from one species to another.[60]
Community ecology emerged from natural history and population biology. Not only does it include the study of the interactions between species, but it also focuses on ecological concepts such as mutualism, predation, and competition within communities.[61] It is used to explicate properties such as diversity, dominance, and composition of a community.[62] There are three primary approaches to integrating phylogenetic information into studies of community organizations. The first approach focuses on examining the phylogenetic structure of community assemblages. The second approach focuses on exploring the phylogenetic basis of community niche structures. The final way zones in on adding a community context to studies of trait evolution and biogeography.[57]
Species concepts
Species concepts are the subject of debate in the field of molecular ecology. Since the beginning of taxonomy, scientists have wanted to standardize and perfect the way species are defined. There are many species concepts that dictate how ecologists determine a good species. The most commonly used concept is the biological species concept which defines a species as groups of actually or potentially interbreeding natural populations, which are reproductively isolated from other such groups (Mayr, 1942).[58] This concept is not always useful, particularly when it comes to hybrids. Other species concepts include phylogenetic species concept which describes a species as the smallest identifiable monophyletic group of organisms within which there is a parental pattern of ancestry and descent.[58] This concept defines species on the identifiable. It would also suggest that until two identifiable groups actually produce offspring, they remain separate species. In 1999, John Avise and Glenn Johns suggested a standardized method for defining species based on past speciation and measuring biological classifications as time dependent. Their method used temporal banding to make genus, family and order based on how many tens of millions of years ago the speciation event that resulted in each species took place.[63]


Landscape genetics
Landscape genetics is a rapidly emerging interdisciplinary field within molecular ecology. Landscape genetics relates genetics to landscape characteristics, such as land-cover use (forests, agriculture, roads, etc.), presence of barriers, and corridors, rivers, elevation, etc. Landscape genetics answers how landscape affects dispersal and gene flow.
Barriers are any landscape features that prevents dispersal.[27] Barriers for terrestrial species can include mountains, rivers, roads, and unsuitable terrain, such as agriculture fields. Barriers for aquatic species can include islands or dams. Barriers are species specific; for example a river is a barrier to a field mouse, while a hawk can fly over a river. Corridors are areas over which dispersal is possible.[27] Corridors are stretches of suitable habitat and can also be man-made, such as overpasses over roads and fish ladders on dams.
Geographic data used for landscape genetics can include data collected by radars in planes, land satellite data, marine data collected by NOAA, as well as any other ecological data. In landscape genetics researchers often use different analyses to attempt to determine the best way for a species to travel from point A to point B. Least cost path analysis uses geographic data to determine the most efficient path from one point to another.[27]:131–337–341 Circuit scape analysis predicts all the possible paths and the probability of each path's use between point A and point B. These analyses are used to determine the route a dispersing individual is likely to travel.
Landscape genetics is becoming an increasingly important tool in wildlife conservation efforts. It is being used to determine how habitat loss and fragmentation affects the movement of species.[64] It is also used to determine which species need to be managed and whether to manage subpopulations the same or differently according to their gene flow.
See also
References
- Charlesworth B. "Measures of Divergence Between Populations and the Effect of Forces that Reduce Variability" (PDF). Cite journal requires
|journal=(help) - Miglani (2015). Essentials of Molecular Genetics. Alpha Science International Limited. p. 36.
- Freeland JR (2014). "Molecular Ecology". eLS. John Wiley & Sons, Inc. doi:10.1002/9780470015902.a0003268.pub2. ISBN 978-0-470-01590-2.
- Lauber CL, Hamady M, Knight R, Fierer N (August 2009). "Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale". Applied and Environmental Microbiology. 75 (15): 5111–20. doi:10.1128/AEM.00335-09. PMC 2725504. PMID 19502440.
- Větrovský T, Baldrian P (2013). "Analysis of soil fungal communities by amplicon pyrosequencing: current approaches to data analysis and the introduction of the pipeline SEED". Biology and Fertility of Soils. 49 (8): 1027–1037. doi:10.1007/s00374-013-0801-y. S2CID 14072170.
- Amend AS, Seifert KA, Samson R, Bruns TD (August 2010). "Indoor fungal composition is geographically patterned and more diverse in temperate zones than in the tropics". Proceedings of the National Academy of Sciences of the United States of America. 107 (31): 13748–53. Bibcode:2010PNAS..10713748A. doi:10.1073/pnas.1000454107. PMC 2922287. PMID 20616017.
- Buée M, Reich M, Murat C, Morin E, Nilsson RH, Uroz S, Martin F (October 2009). "454 Pyrosequencing analyses of forest soils reveal an unexpectedly high fungal diversity". The New Phytologist. 184 (2): 449–56. doi:10.1111/j.1469-8137.2009.03003.x. PMID 19703112.
- Baldrian P, Kolařík M, Stursová M, Kopecký J, Valášková V, Větrovský T, et al. (February 2012). "Active and total microbial communities in forest soil are largely different and highly stratified during decomposition". The ISME Journal. 6 (2): 248–58. doi:10.1038/ismej.2011.95. PMC 3260513. PMID 21776033.
- Voříšková J, Baldrian P (March 2013). "Fungal community on decomposing leaf litter undergoes rapid successional changes". The ISME Journal. 7 (3): 477–86. doi:10.1038/ismej.2012.116. PMC 3578564. PMID 23051693.
- Seifert KA (May 2009). "Progress towards DNA barcoding of fungi". Molecular Ecology Resources. 9 Suppl s1: 83–9. doi:10.1111/j.1755-0998.2009.02635.x. PMID 21564968.
- Horton TR, Bruns TD (August 2001). "The molecular revolution in ectomycorrhizal ecology: peeking into the black-box". Molecular Ecology. 10 (8): 1855–71. doi:10.1046/j.0962-1083.2001.01333.x. PMID 11555231.
- Weber CF, Vilgalys R, Kuske CR (2013). "Changes in Fungal Community Composition in Response to Elevated Atmospheric CO2 and Nitrogen Fertilization Varies with Soil Horizon". Frontiers in Microbiology. 4: 78. doi:10.3389/fmicb.2013.00078. PMC 3621283. PMID 23641237.
- Kiss L (July 2012). "Limits of nuclear ribosomal DNA internal transcribed spacer (ITS) sequences as species barcodes for Fungi". Proceedings of the National Academy of Sciences of the United States of America. 109 (27): E1811, author reply E1812. Bibcode:2012PNAS..109E1812S. doi:10.1073/pnas.1207508109. PMC 3390861.
- Porras-Alfaro A, Liu KL, Kuske CR, Xie G (February 2014). "From genus to phylum: large-subunit and internal transcribed spacer rRNA operon regions show similar classification accuracies influenced by database composition". Applied and Environmental Microbiology. 80 (3): 829–40. doi:10.1128/AEM.02894-13. PMC 3911224. PMID 24242255.
- Kõljalg U, Larsson KH, Abarenkov K, Nilsson RH, Alexander IJ, Eberhardt U, et al. (June 2005). "UNITE: a database providing web-based methods for the molecular identification of ectomycorrhizal fungi". The New Phytologist. 166 (3): 1063–8. doi:10.1111/j.1469-8137.2005.01376.x. PMID 15869663.
- Liu KL, Porras-Alfaro A, Kuske CR, Eichorst SA, Xie G (March 2012). "Accurate, rapid taxonomic classification of fungal large-subunit rRNA genes". Applied and Environmental Microbiology. 78 (5): 1523–33. doi:10.1128/aem.06826-11. PMC 3294464. PMID 22194300.
- Vilgalys R (2003). "Taxonomic misidentification in public DNA databases". New Phytol. 160: 4–5. doi:10.1046/j.1469-8137.2003.00894.x.
- Nilsson RH, Tedersoo L, Abarenkov K, Ryberg M, Kristiansson E, Hartmann M, et al. (2012). "Five simple guidelines for establishing basic authenticity and reliability of newly generated fungal ITS sequences". MycoKeys. 4: 37–63. doi:10.3897/mycokeys.4.3606.
- Kõljalg U, Nilsson RH, Abarenkov K, Tedersoo L, Taylor AF, Bahram M, et al. (November 2013). "Towards a unified paradigm for sequence-based identification of fungi" (PDF). Molecular Ecology. 22 (21): 5271–7. doi:10.1111/mec.12481. hdl:10261/130958. PMID 24112409.
- Lindahl BD, Nilsson RH, Tedersoo L, Abarenkov K, Carlsen T, Kjøller R, et al. (July 2013). "Fungal community analysis by high-throughput sequencing of amplified markers--a user's guide". The New Phytologist. 199 (1): 288–99. doi:10.1111/nph.12243. PMC 3712477. PMID 23534863.
- Nilsson RH, Kristiansson E, Ryberg M, Hallenberg N, Larsson KH (May 2008). "Intraspecific ITS variability in the kingdom fungi as expressed in the international sequence databases and its implications for molecular species identification". Evolutionary Bioinformatics Online. 4: 193–201. doi:10.4137/EBO.S653. PMC 2614188. PMID 19204817.
- Griffith SC, Owens IP, Thuman KA (November 2002). "Extra pair paternity in birds: a review of interspecific variation and adaptive function". Molecular Ecology. 11 (11): 2195–212. doi:10.1046/j.1365-294X.2002.01613.x. PMID 12406233.
- Hunt J, Simmons LW (2002-09-01). "Confidence of paternity and paternal care: covariation revealed through the experimental manipulation of the mating system in the beetle Onthophagus taurus". Journal of Evolutionary Biology. 15 (5): 784–795. doi:10.1046/j.1420-9101.2002.00442.x.
- Neff BD (April 2003). "Decisions about parental care in response to perceived paternity". Nature. 422 (6933): 716–9. Bibcode:2003Natur.422..716N. doi:10.1038/nature01528. PMID 12700761. S2CID 3861679.
- Conrad KF, Johnston PV, Crossman C, Kempenaers B, Robertson RJ, Wheelwright NT, Boag PT (May 2001). "High levels of extra-pair paternity in an isolated, low-density, island population of tree swallows (Tachycineta bicolor)". Molecular Ecology. 10 (5): 1301–8. doi:10.1046/j.1365-294X.2001.01263.x. PMID 11380885.
- Kempenaers B, Everding S, Bishop C, Boag P, Robertson RJ (2001-01-18). "Extra-pair paternity and the reproductive role of male floaters in the tree swallow (Tachycineta bicolor)". Behavioral Ecology and Sociobiology. 49 (4): 251–259. doi:10.1007/s002650000305. ISSN 0340-5443. S2CID 25483760.
- Freeland J, Kirk H, Petersen S (2011). Molecular Ecology. West Sussex, UK: John Wiley & Sons, Ltd. p. 295. ISBN 978-1-119-99308-7.
- Schwarzová L, Šimek J, Coppack T, Tryjanowski P (2008-06-01). "Male-Biased Sex of Extra Pair Young in the Socially Monogamous Red-Backed Shrike Lanius collurio". Acta Ornithologica. 43 (2): 235–239. doi:10.3161/000164508X395379. ISSN 0001-6454. S2CID 84293669.
- Johnson LS, Thompson CF, Sakaluk SK, Neuhäuser M, Johnson BG, Soukup SS, et al. (June 2009). "Extra-pair young in house wren broods are more likely to be male than female". Proceedings. Biological Sciences. 276 (1665): 2285–9. doi:10.1098/rspb.2009.0283. PMC 2677618. PMID 19324727.
- Wright S (March 1943). "Isolation by Distance". Genetics. 28 (2): 114–38. PMC 1209196. PMID 17247074.
- Jenkins DG, Carey M, Czerniewska J, Fletcher J, Hether T, Jones A, et al. (2010). "A meta-analysis of isolation by distance: relic or reference standard for landscape genetics?". Ecography. 33: 315–320. doi:10.1111/j.1600-0587.2010.06285.x.
- Levins R (1969). "Some demographic and genetic consequences of environmental heterogeneity for biological control". Bulletin of the Entomological Society of America. 15 (3): 237–240. doi:10.1093/besa/15.3.237.
- Brown WM, Prager EM, Wang A, Wilson AC (1982). "Mitochondrial DNA sequences of primates: tempo and mode of evolution" (PDF). Journal of Molecular Evolution. 18 (4): 225–39. Bibcode:1982JMolE..18..225B. doi:10.1007/bf01734101. hdl:2027.42/48036. PMID 6284948.
- Kumar S, Hedges SB (April 1998). "A molecular timescale for vertebrate evolution". Nature. 392 (6679): 917–20. Bibcode:1998Natur.392..917K. doi:10.1038/31927. PMID 9582070. S2CID 205001573.
- Yasui Y (March 1997). "A "Good-Sperm" model can explain the evolution of costly multiple mating by females". The American Naturalist. 149 (3): 573–584. doi:10.1086/286006.
- Puurtinen M, Ketola T, Kotiaho JS (April 2005). "Genetic compatibility and sexual selection". Trends in Ecology & Evolution. 20 (4): 157–8. doi:10.1016/j.tree.2005.02.005. PMID 16701361.
- Wedekind C, Seebeck T, Bettens F, Paepke AJ (June 1995). "MHC-dependent mate preferences in humans". Proceedings. Biological Sciences. 260 (1359): 245–9. Bibcode:1995RSPSB.260..245W. doi:10.1098/rspb.1995.0087. PMID 7630893. S2CID 34971350.
- Végvári Z, Katona G, Vági B, Freckleton RP, Gaillard JM, Székely T, Liker A (July 2018). "Sex-biased breeding dispersal is predicted by social environment in birds". Ecology and Evolution. 8 (13): 6483–6491. doi:10.1002/ece3.4095. PMC 6053579. PMID 30038750.
- Hjernquist MB, Thuman Hjernquist KA, Forsman JT, Gustafsson L (2009-03-01). "Sex allocation in response to local resource competition over breeding territories". Behavioral Ecology. 20 (2): 335–339. doi:10.1093/beheco/arp002. ISSN 1045-2249.
- Herre EA (May 1985). "Sex ratio adjustment in fig wasps". Science. 228 (4701): 896–8. Bibcode:1985Sci...228..896H. doi:10.1126/science.228.4701.896. PMID 17815055.
- Verkuil YI, Juillet C, Lank DB, Widemo F, Piersma T (September 2014). "Genetic variation in nuclear and mitochondrial markers supports a large sex difference in lifetime reproductive skew in a lekking species". Ecology and Evolution. 4 (18): 3626–32. doi:10.1002/ece3.1188. PMC 4224536. PMID 25478153.
- Zhang L, Qu J, Li K, Li W, Yang M, Zhang Y (October 2017). "Genetic diversity and sex-bias dispersal of plateau pika in Tibetan plateau". Ecology and Evolution. 7 (19): 7708–7718. doi:10.1002/ece3.3289. PMC 5632614. PMID 29043027.
- "Spatial autocorrelation — R Spatial". rspatial.org. Retrieved 2020-04-03.
- Antoniazza S, Burri R, Fumagalli L, Goudet J, Roulin A (July 2010). "Local adaptation maintains clinal variation in melanin-based coloration of European barn owls (Tyto alba)". Evolution; International Journal of Organic Evolution. 64 (7): 1944–54. doi:10.1111/j.1558-5646.2010.00969.x. PMID 20148951.
- Woiwood I, Reynolds DR, Thomas CD (2001). Insect Movement: Mechanisms and Consequences : Proceedings of the Royal Entomological Society's 20th Symposium. CABI. ISBN 978-0-85199-781-0.
- "Inbreeding and Population Structure" (PDF). University of Vermont.
- Hu XS, He F (July 2005). "Background selection and population differentiation" (PDF). Journal of Theoretical Biology. 235 (2): 207–19. doi:10.1016/j.jtbi.2005.01.004. PMID 15862590.
- "An Introduction to Restoration Genetics: How is Genetic Diversity Distributed in Natural Populations?". www.nps.gov.
- Charlesworth D, Willis JH (November 2009). "The genetics of inbreeding depression". Nature Reviews. Genetics. 10 (11): 783–96. doi:10.1038/nrg2664. PMID 19834483. S2CID 771357.
- Charlesworth, Deborah; Willis, John H. (November 2009). "The genetics of inbreeding depression". Nature Reviews Genetics. 10 (11): 783–796. doi:10.1038/nrg2664. ISSN 1471-0064. PMID 19834483.
- Frankham R, Ballou JD, Eldridge MD, Lacy RC, Ralls K, Dudash MR, Fenster CB (June 2011). "Predicting the probability of outbreeding depression". Conservation Biology. 25 (3): 465–75. doi:10.1111/j.1523-1739.2011.01662.x. PMID 21486369.
- Turcek FJ, Hickey JJ (January 1951). "Effect of Introductions on Two Game Populations in Czechoslovakia". The Journal of Wildlife Management. 15 (1): 113. doi:10.2307/3796784. JSTOR 3796784.
- Fenster CB, Galloway LF (2000-10-18). "Inbreeding and Outbreeding Depression in Natural Populations of Chamaecrista fasciculata (Fabaceae)". Conservation Biology. 14 (5): 1406–1412. doi:10.1046/j.1523-1739.2000.99234.x. ISSN 0888-8892.
- Frankham R, Ballou JD, Briscoe DA, McInness KH (2010). Introduction to conservation genetics (2nd ed.). Cambridge, UK: Cambridge University Press. ISBN 978-0-521-87847-0. OCLC 268793768.
- Freeland J, Kirk H, Petersen S (2011). Molecular Ecology. Chichester, West Sussex, UK: John Wiley & Sons. pp. 332–333. ISBN 978-0-470-74833-6.
- Vandersteen Tymchuk W, O'Reilly P, Bittman J, Macdonald D, Schulte P (May 2010). "Conservation genomics of Atlantic salmon: variation in gene expression between and within regions of the Bay of Fundy". Molecular Ecology. 19 (9): 1842–59. doi:10.1111/j.1365-294X.2010.04596.x. PMID 20529070.
- Webb CO, Ackerly DD, McPeek MA, Donoghue MJ (November 2002). "Phylogenies and Community Ecology". Annual Review of Ecology and Systematics. 33 (1): 475–505. doi:10.1146/annurev.ecolsys.33.010802.150448. ISSN 0066-4162.
- Freeland J, Petersen S, Kirk H (2011). Molecular Ecology (2nd ed.). Chichester, West Sussex, UK: Wiley-Blackwell.
- "Reading trees: A quick review". Understanding Evolution. University of California Museum of Paleontology. 22 August 2008.
- Harrison, Larson, Richard, Erica (2014). "Hybridization, introgression, and the nature of species boundaries". Journal of Heredity. 105.
- "Community ecology - Latest research and news | Nature". www.nature.com. Retrieved 2020-03-07.
- Jackson ST, Blois JL (April 2015). "Community ecology in a changing environment: Perspectives from the Quaternary". Proceedings of the National Academy of Sciences of the United States of America. 112 (16): 4915–21. Bibcode:2015PNAS..112.4915J. doi:10.1073/pnas.1403664111. PMC 4413336. PMID 25901314.
- Avise JC, Johns GC (June 1999). "Proposal for a standardized temporal scheme of biological classification for extant species". Proceedings of the National Academy of Sciences of the United States of America. 96 (13): 7358–63. Bibcode:1999PNAS...96.7358A. doi:10.1073/pnas.96.13.7358. PMC 22090. PMID 10377419.
- Holderegger R, Wagner HH (2008-03-01). "Landscape Genetics". BioScience. 58 (3): 199–207. doi:10.1641/B580306.