Mixed quantum-classical dynamics
Mixed quantum-classical (MQC) dynamics is a class of computational theoretical chemistry methods tailored to simulate nonadiabatic (NA) processes in molecular and supramolecular chemistry.[1] Such methods are characterized by:
- Propagation of nuclear dynamics through classical trajectories;
- Propagation of the electrons (or fast particles) through quantum methods;
- A feedback algorithm between the electronic and nuclear subsystems to recover nonadiabatic information.
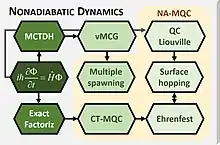
Use of NA-MQC dynamics
In the Born-Oppenheimer approximation, the ensemble of electrons of a molecule or supramolecular system can have several discrete states. The potential energy of each of these electronic states depends on the position of the nuclei, forming multidimensional surfaces.
Under usual conditions (room temperature, for instance), the molecular system is in the ground electronic state (the electronic state of lowest energy). In this stationary situation, nuclei and electrons are in equilibrium, and the molecule naturally vibrates near harmonically due to the zero-point energy.
Particle collisions and photons with wavelengths in the range from visible to X-ray can promote the electrons to electronically excited states. Such events create a non-equilibrium between nuclei and electrons, which leads to an ultrafast response (picosecond scale) of the molecular system. During the ultrafast evolution, the nuclei may reach geometric configurations where the electronic states mix, allowing the system to transfer to another state spontaneously. These state transfers are nonadiabatic phenomena.
Nonadiabatic dynamics is the field of computational chemistry that simulates such ultrafast nonadiabatic response.
In principle, the problem can be exactly addressed by solving the time-dependent Schrödinger equation (TDSE) for all particles (nuclei and electrons). Methods like the multiconfigurational self-consistent Hartree (MCTDH) have been developed to do such task.[2] Nevertheless, they are limited to small systems with two dozen degrees of freedom due to the enormous difficulties of developing multidimensional potential energy surfaces and the costs of the numerical integration of the quantum equations.
NA-MQC dynamics methods have been developed to reduce the burden of these simulations by profiting from the fact that the nuclear dynamics is near classical.[3] Treating the nuclei classically allows simulating the molecular system in full dimensionality. The impact of the underlying assumptions depends on each particular NA-MQC method.
Most of NA-MQC dynamics methods have been developed to simulate internal conversion (IC), the nonadiabatic transfer between states of the same spin multiplicity. The methods have been extended, however, to deal with other types of processes like intersystem crossing (ISC; transfer between states of different multiplicities)[4] and field-induced transfers.[5]
NA-MQC dynamics has been often used in theoretical investigations of photochemistry and femtochemistry, especially when time-resolved processes are relevant.[6][7]
List of NA-MQC dynamics methods
NA-MQC dynamics is a general class of methods developed since the 1970s. It encompasses:
- Trajectory surface hopping (TSH; FSSH for fewest switches surface hopping);[8]
- Multiple spawning (AIMS for ab initio multiple spawning; FMS for full multiple spwaning);[9]
- Mean-field Ehrenfest dynamics (MFE);[3]
- Coupled-Trajectory Mixed Quantum-Classical Algorithm (CT-MQC);[10]
- Mixed quantum−classical Liouville equation (QCLE);[11]
- Mapping approach;[12]
- Nonadiabatic Bohmian dynamics (NABDY).[13]
- Multiple cloning (AIMC for ab initio multiple cloning)[14]
Integration of NA-MQC dynamics
Classical trajectories
The classical trajectories can be integrated with conventional methods, as the Verlet algorithm. Such integration requires the forces acting on the nuclei. They are proportional to the gradient of the potential energy of the electronic states and can be efficiently computed with diverse electronic structure methods for excited states, like the multireference configuration interaction (MRCI) or the linear-response time-dependent density functional theory (TDDFT).
In NA-MQC methods like FSSH or MFE, the trajectories are independent of each other. In such a case, they can be separately integrated and only grouped afterward for the statistical analysis of the results. In methods like CT-MQC or diverse TSH variants,[15] the trajectories are coupled and must be integrated simultaneously.
Electronic subsystem
In NA-MQC dynamics, the electrons are usually treated by a local approximation of the TDSE, i.e., they depend only on the electronic forces and couplings at the instantaneous position of the nuclei.
Nonadiabatic algorithms

There are three basic algorithms to recover nonadiabatic information in NA-MQC methods:[1]
- Spawning - new trajectories are created at regions of large nonadiabatic coupling.
- Hopping - trajectories are propagated on a single potential energy surface (PES), but they are allowed to change surface near regions of large nonadiabatic couplings.
- Averaging - trajectories are propagated on a weighted average of potential energy surfaces. The weights are determined by the amount of nonadiabatic mixing.
Relation to other nonadiabatic methods
NA-MQC dynamics are approximated methods to solve the time-dependent Schrödinger equation for a molecular system. Methods like TSH, in particular in the fewest switches surface hopping (FSSH) formulation, do not have an exact limit.[16] Other methods like MS or CT-MQC can in principle deliver the exact non-relativistic solution.[9][10]
In the case of multiple spawning, it is hierarchically connected to MCTDH,[2] while CT-MQC is connected to the exact factorization method.[10]
Drawbacks in NA-MQC dynamics
The most common approach in NA-MQC dynamics is to compute the electronic properties on-the-fly, i.e., at each timestep of the trajectory integration. Such an approach has the advantage of not requiring pre-computed multidimensional potential energy surfaces. Nevertheless, the costs associated with the on-the-fly approach are significantly high, leading to a systematic level downgrade of the simulations. This downgrade has been shown to lead to qualitatively wrong results.[17]
The local approximation implied by the classical trajectories in NA-MQC dynamics also leads to failing in the description of non-local quantum effects, as tunneling and quantum interference. Some methods like MFE and FSSH are also affected by decoherence errors.[18] New algorithms have been developed to include tunneling[19] and decoherence effects.[20][21] Global quantum effects can also be considered by applying quantum forces between trajectories.[10]
Software for NA-MQC dynamics
Survey of NA-MQC dynamics implementations in public software.
| Program | Electronic structure methods | NA-MQC method |
| Dedicated NA-MQC dynamics software | ||
| Ant | analytical PES | FSSH, FSTU, FSTU/SD, CSDM, MFE, army ant tunnelling |
| Cobramm | MCSCF, MRCI/OMx, QM/MM | FSSH |
| DFTBaby | TD-(LC)-DFTB
FSSH | |
| Jade | LR-TDDFT, CIS, ADC(2) | FSSH |
| Libra | Analytical PES | FSSH, GFSH, MSSH, MFE (external fields) |
| Na-esmd | CEO, TDHF/semiempirical, CIS/semiempirical | FSSH |
| Newton-X | MRCI, MR-AQCC, MCSCF, ADC(2), CC2, CIS, LR-TDDFT, XMS-CASPT2,a TD-DFTB,a QM/MM, analytical PES, user-defined PES | FSSH (IC and ISCa) |
| Pyxaid | RT-TDKS, RT-SCC-DFTB | FSSH, DISH (external fields) |
| Sharc | MCSCF, MRCI, MS-CASPT2, ADC(2), LR-TDDFT, analytical PES, vibronic coupling models, Frenkel exciton modela | FSSH, SHARC |
| Electronic structure software with NA-MQC options | ||
| Cpmd | LR-TDDFT, ROKS, QM/MM | FSSH, MFE, CT-MQCa (IC and ISC) |
| Gamessa | CASSCF | AIMS |
| Gpawa | RT-TDKS | MFE |
| ChemShella | MRCI/OMx | FSSH |
| Molcas | SA-CASSCF | FSSH |
| Molpro | CASSCF, MS-CASPT2 | AIMS |
| Mopaca | FOMO-CI | FSSH and AIMS (IC and ISC) |
| Octopus | RT-TDKS | MFE |
| Turbomole | LR-TDDFT | FSSH |
| Q-Chem | LR-TDDFT, CIS | FSSH, A-FSSH |
a Development version.
References
- Crespo-Otero, Rachel; Barbatti, Mario (16 May 2018). "Recent Advances and Perspectives on Nonadiabatic Mixed Quantum–Classical Dynamics" (PDF). Chemical Reviews. 118 (15): 7026–7068. doi:10.1021/acs.chemrev.7b00577. PMID 29767966.
- Worth, Graham A.; Hunt, Patricia; Robb, Michael A. (February 2003). "Nonadiabatic Dynamics: A Comparison of Surface Hopping Direct Dynamics with Quantum Wavepacket Calculations". The Journal of Physical Chemistry A. 107 (5): 621–631. Bibcode:2003JPCA..107..621W. doi:10.1021/jp027117p.
- Tully, John C. (1998). "Mixed quantum–classical dynamics". Faraday Discussions. 110: 407–419. Bibcode:1998FaDi..110..407T. doi:10.1039/A801824C.
- Granucci, Giovanni; Persico, Maurizio; Spighi, Gloria (14 December 2012). "Surface hopping trajectory simulations with spin-orbit and dynamical couplings". The Journal of Chemical Physics. 137 (22): 22A501. doi:10.1063/1.4707737. PMID 23249038.
- Mitrić, Roland; Petersen, Jens; Wohlgemuth, Matthias; Werner, Ute; Bonačić-Koutecký, Vlasta; Wöste, Ludger; Jortner, Joshua (28 April 2011). "Time-Resolved Femtosecond Photoelectron Spectroscopy by Field-Induced Surface Hopping". The Journal of Physical Chemistry A. 115 (16): 3755–3765. doi:10.1021/jp106355n. PMID 20939619.
- Akimov, Alexey V.; Prezhdo, Oleg V. (8 April 2015). "Large-Scale Computations in Chemistry: A Bird's Eye View of a Vibrant Field". Chemical Reviews. 115 (12): 5797–5890. doi:10.1021/cr500524c.
- Brunk, Elizabeth; Rothlisberger, Ursula (16 April 2015). "Mixed Quantum Mechanical/Molecular Mechanical Molecular Dynamics Simulations of Biological Systems in Ground and Electronically Excited States". Chemical Reviews. 115 (12): 6217–6263. doi:10.1021/cr500628b. PMID 25880693.
- Tully, John C. (15 July 1990). "Molecular dynamics with electronic transitions". The Journal of Chemical Physics. 93 (2): 1061–1071. Bibcode:1990JChPh..93.1061T. doi:10.1063/1.459170.
- Curchod, Basile F. E.; Martínez, Todd J. (21 February 2018). "Ab Initio Nonadiabatic Quantum Molecular Dynamics" (PDF). Chemical Reviews. 118 (7): 3305–3336. doi:10.1021/acs.chemrev.7b00423. PMID 29465231.
- Agostini, Federica; Min, Seung Kyu; Abedi, Ali; Gross, E. K. U. (19 April 2016). "Quantum-Classical Nonadiabatic Dynamics: Coupled- vs Independent-Trajectory Methods". Journal of Chemical Theory and Computation. 12 (5): 2127–2143. arXiv:1512.04638. doi:10.1021/acs.jctc.5b01180. PMID 27030209.
- Kapral, Raymond; Ciccotti, Giovanni (8 May 1999). "Mixed quantum-classical dynamics". The Journal of Chemical Physics. 110 (18): 8919–8929. Bibcode:1999JChPh.110.8919K. doi:10.1063/1.478811.
- Thoss, Michael; Stock, Gerhard (January 1999). "Mapping approach to the semiclassical description of nonadiabatic quantum dynamics". Physical Review A. 59 (1): 64–79. Bibcode:1999PhRvA..59...64T. doi:10.1103/PhysRevA.59.64.
- Curchod, Basile F. E.; Tavernelli, Ivano (2013-05-14). "On trajectory-based nonadiabatic dynamics: Bohmian dynamics versus trajectory surface hopping". The Journal of Chemical Physics. 138 (18): 184112. doi:10.1063/1.4803835. ISSN 0021-9606.
- Makhov, Dmitry V.; Glover, William J.; Martinez, Todd J.; Shalashilin, Dmitrii V. (7 August 2014). "multiple cloning algorithm for quantum nonadiabatic molecular dynamics". The Journal of Chemical Physics. 141 (5): 054110. doi:10.1063/1.4891530.
- Wang, Linjun; Akimov, Alexey; Prezhdo, Oleg V. (23 May 2016). "Recent Progress in Surface Hopping: 2011–2015". The Journal of Physical Chemistry Letters. 7 (11): 2100–2112. doi:10.1021/acs.jpclett.6b00710.
- Ou, Qi; Subotnik, Joseph E. (19 September 2013). "Electronic Relaxation in Benzaldehyde Evaluated via TD-DFT and Localized Diabatization: Intersystem Crossings, Conical Intersections, and Phosphorescence". The Journal of Physical Chemistry C. 117 (39): 19839–19849. doi:10.1021/jp405574q.
- Plasser, Felix; Crespo-Otero, Rachel; Pederzoli, Marek; Pittner, Jiri; Lischka, Hans; Barbatti, Mario (13 March 2014). "Surface Hopping Dynamics with Correlated Single-Reference Methods: 9H-Adenine as a Case Study". Journal of Chemical Theory and Computation. 10 (4): 1395–1405. doi:10.1021/ct4011079. PMID 26580359.
- Subotnik, Joseph E.; Jain, Amber; Landry, Brian; Petit, Andrew; Ouyang, Wenjun; Bellonzi, Nicole (27 May 2016). "Understanding the Surface Hopping View of Electronic Transitions and Decoherence". Annual Review of Physical Chemistry. 67 (1): 387–417. doi:10.1146/annurev-physchem-040215-112245. PMID 27215818.
- Zheng, Jingjing; Xu, Xuefei; Meana-Pañeda, Rubén; Truhlar, Donald G. (2014). "Army ants tunneling for classical simulations". Chem. Sci. 5 (5): 2091–2099. doi:10.1039/C3SC53290A.
- Granucci, Giovanni; Persico, Maurizio; Zoccante, Alberto (7 October 2010). "Including quantum decoherence in surface hopping". The Journal of Chemical Physics. 133 (13): 134111. doi:10.1063/1.3489004. PMID 20942527.
- Jain, Amber; Alguire, Ethan; Subotnik, Joseph E. (7 October 2016). "An Efficient, Augmented Surface Hopping Algorithm That Includes Decoherence for Use in Large-Scale Simulations". Journal of Chemical Theory and Computation. 12 (11): 5256–5268. doi:10.1021/acs.jctc.6b00673. PMID 27715036.