List of protein structure prediction software
This list of protein structure prediction software summarizes notable used software tools in protein structure prediction, including homology modeling, protein threading, ab initio methods, secondary structure prediction, and transmembrane helix and signal peptide prediction.
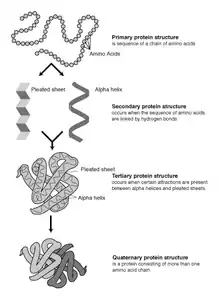
Constituent amino-acids can be analyzed to predict secondary, tertiary and quaternary protein structure.
Software list
Below is a list which separates programs according to the method used for structure prediction.
Homology modeling
| Name | Method | Description | Link |
|---|---|---|---|
| RaptorX | remote homology detection, protein 3D modeling, binding site prediction | Automated webserver and Downloadable program | server and download |
| Biskit | wraps external programs into automated workflow | BLAST search, T-Coffee alignment, and MODELLER construction | project site |
| ESyPred3D | Template detection, alignment, 3D modeling | Automated webserver | server |
| FoldX | Energy calculations and protein design | Downloadable program | download |
| Phyre and Phyre2 | Remote template detection, alignment, 3D modeling, multi-templates, ab initio | Webserver with job manager, automatically updated fold library, genome searching and other facilities | server |
| HHpred | Template detection, alignment, 3D modeling | Interactive webserver with help facility | server download article |
| MODELLER | Satisfaction of spatial restraints | Standalone program mainly in Fortran and Python | download Server |
| CONFOLD | Satisfaction of contact and distance restraints | Standalone program mainly in Fortran and Perl | download |
| MOE (Molecular Operating Environment) | Template identification, use of multiple templates and accounting for other environments (e.g. excluded ligand volumes), loop modelling, rotamer libraries for sidechain conformations, relaxation using MM forcefields. | Proprietary platform, supported on Windows, Linux and Mac | site |
| ROBETTA | Rosetta homology modeling and ab initio fragment assembly with Ginzu domain prediction | Webserver | server |
| BHAGEERATH-H | Combination of ab initio folding and homology methods | Protein tertiary structure predictions | server |
| SWISS-MODEL | Local similarity/fragment assembly | Automated webserver (based on ProModII) | server |
| Yasara | Detection of templates, alignment, modeling incl. ligands and oligomers, hybridization of model fragments | Graphical interface or text mode (clusters) | Home page CASP8 results |
| AWSEM-Suite | Molecular dynamics simulation based on template-guided, coevolutionary-enhanced optimized folding landscapes[1] | Automated webserver | server |
Threading/fold recognition
| Name | Method | Description | Link |
|---|---|---|---|
| RaptorX | Remote template detection, single-template and multi-template threading, totally different from and much better than the old program RAPTOR designed by the same group | Webserver with job manager, automatically updated fold library | download server |
| HHpred | Template detection, alignment, 3D modeling | Interactive webserver with help facility | server download article |
| Phyre and Phyre2 | Remote template detection, alignment, 3D modeling, multi-templates, ab initio | Webserver with job manager, automatically updated fold library, genome searching and other facilities | server |
Ab initio structure prediction
| Name | Method | Description | Link |
|---|---|---|---|
| trRosetta | trRosetta is an algorithm for fast and accurate de novo protein structure prediction. It builds the protein structure based on direct energy minimizations with a restrained Rosetta. The restraints include inter-residue distance and orientation distributions, predicted by a deep residual neural network. | Webserver and source codes. It takes about one hour to fold proteins with ~300 AAs | Server |
| I-TASSER | Threading fragment structure reassembly | On-line server for protein modeling | Server |
| ROBETTA | Rosetta homology modeling and ab initio fragment assembly with Ginzu domain prediction | Webserver | server |
| Rosetta@home | Distributed-computing implementation of Rosetta algorithm | Downloadable program | main page |
| Abalone | Molecular Dynamics folding | Program | Example |
Secondary structure prediction
Detailed list of programs can be found at List of protein secondary structure prediction programs
See also
- List of protein secondary structure prediction programs
- Comparison of nucleic acid simulation software
- List of software for molecular mechanics modeling
- Molecular design software
- Protein design
External link
- bio.tools, finding more tools
References
- Jin, Shikai; Contessoto, Vinicius G; Chen, Mingchen; Schafer, Nicholas P; Lu, Wei; Chen, Xun; Bueno, Carlos; Hajitaheri, Arya; Sirovetz, Brian J; Davtyan, Aram; Papoian, Garegin A; Tsai, Min-Yeh; Wolynes, Peter G (2 July 2020). "AWSEM-Suite: a protein structure prediction server based on template-guided, coevolutionary-enhanced optimized folding landscapes". Nucleic Acids Research. 48 (W1): W25–W30. doi:10.1093/nar/gkaa356. PMC 7319565. PMID 32383764.
This article is issued from Wikipedia. The text is licensed under Creative Commons - Attribution - Sharealike. Additional terms may apply for the media files.