Lewis acid catalysis
In Lewis acid catalysis of organic reactions, a metal-based Lewis acid acts as an electron pair acceptor to increase the reactivity of a substrate. Common Lewis acid catalysts are based on main group metals such as aluminum, boron, silicon, and tin, as well as many early (titanium, zirconium) and late (iron, copper, zinc) d-block metals. The metal atom forms an adduct with a lone-pair bearing electronegative atom in the substrate, such as oxygen (both sp2 or sp3), nitrogen, sulfur, and halogens. The complexation has partial charge-transfer character and makes the lone-pair donor effectively more electronegative, activating the substrate toward nucleophilic attack, heterolytic bond cleavage, or cycloaddition with 1,3-dienes and 1,3-dipoles.[1]

Many classical reactions involving carbon–carbon or carbon–heteroatom bond formation can be catalyzed by Lewis acids. Examples include the Friedel-Crafts reaction, the aldol reaction, and various pericyclic processes that proceed slowly at room temperature, such as the Diels-Alder reaction and the ene reaction. In addition to accelerating the reactions, Lewis acid catalysts are able to impose regioselectivity and stereoselectivity in many cases.
Early developments in Lewis acid reagents focused on easily available compounds such as TiCl4, BF3, SnCl4, and AlCl3. Over the years, versatile catalysts bearing ligands designed for specific applications have facilitated improvement in both reactivity and selectivity of Lewis acid-catalyzed reactions. More recently, Lewis acid catalysts with chiral ligands have become an important class of tools for asymmetric catalysis.[2]
Challenges in the development of Lewis acid catalysis include inefficient catalyst turnover (caused by catalyst affinity for the product) and the frequent requirement of two-point binding for stereoselectivity, which often necessitates the use of auxiliary groups.
Mechanism
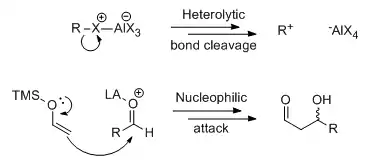
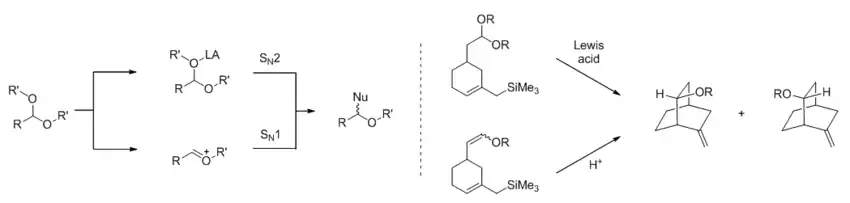
In reactions with polar mechanisms, Lewis acid catalysis often involves binding of the catalyst to Lewis basic heteroatoms and withdrawing electron density, which in turn facilitates heterolytic bond cleavage (in the case of Friedel-Crafts reaction) or directly activates the substrate toward nucleophilic attack (in the case of carbonyl addition reactions). The dichotomy can have important consequences in some reactions, as in the case of Lewis acid-promoted acetal substitution reactions, where the SN1 and SN2 mechanisms shown below may give different stereochemical outcomes. Studying the product ratio in a bicyclic system, Denmark and colleagues showed that both mechanisms could be operative depending on the denticity of the Lewis acid and the identity of the R' group.[3]
In Diels-Alder and 1,3-dipolar cycloaddition reactions, Lewis acids lower the LUMO energy of the dienophile or dipolarphile, respectively, making it more reactive toward the diene or the dipole.
Lewis acid catalysis with carbonyl-containing substrates
Among the types of reactions that can be catalyzed by Lewis acids, those with carbonyl-containing substrates have received the greatest amount of attention. The first major discovery in this area was in 1960, when Yates and Eaton reported the significant acceleration of the Diels-Alder reaction by AlCl3 when maleic anhydride is the dienophile.[4] Early theoretical studies that depended on frontier orbital analysis established that Lewis acid catalysis operates via lowering of the dienophile's LUMO energy,[5] which is still the accepted rationalization. The concept of lowered LUMO energy is also used to explain the dramatically enhanced electrophilic reactivity of carbonyl compounds (whose LUMO is the C-O π* orbital) towards mild nucleophilic reagents, as in the cases of the Mukaiyama aldol reaction and Sakurai reaction.
In addition to rate acceleration, Lewis acid-catalyzed reactions sometimes exhibit enhanced stereoselectivity, which stimulated the development of stereoinduction models. The models have their roots in knowledge of the structures of Lewis acid-carbonyl complexes which, through decades of research in theoretical calculations, NMR spectroscopy, and X-ray crystallography, were fairly firmly established in the early 1990s:[6]
- σ-Complexation: The complex in which the Lewis acid interacts with the carbonyl compound through a σ-bond with the oxygen lone pair is both thermodynamically favored and catalytically relevant.[7]
- Bent geometry: The metal-oxygen-carbon bond angle is less than 180°, and the metal is syn to the smaller substituent, unless influenced by a chelating group on the larger substituent.
- An s-trans preference for α,β-unsaturated compounds.
Addition and conjugate addition to carbonyl compounds
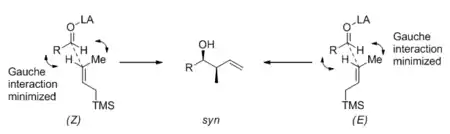
The Mukaiyama aldol reaction and the Sakurai reaction refer to the addition of silyl enol ethers and allylsilanes to carbonyl compounds, respectively. Only under Lewis acid catalysis do these reactions occur under synthetically useful conditions. Acyclic transition states are believed to be operating in both reactions for either 1,2- or 1,4- addition, and steric factors control stereoselectivity. This is in contrast with the rigid Zimmerman-Traxler cyclic transition state that has been widely accepted for the aldol reaction with lithium, boron, and titanium enolates. As a consequence, the double bond geometry in the silyl enol ether or allylsilane does not translate well into product stereochemistry. A model for the Sakurai 1,2-addition, proposed by Kumada, is presented in the scheme below;[8] the syn diastereomer is predominant when the (E) silane is used, and also slightly favored when the (Z) silane is used. A similar analysis by Heathcock[9] explains the fact that, with simple substrates, there is essentially no diastereoselectivity for the intermolecular Mukaiyama aldol reaction.
The Lewis acid catalyst plays a role in stereoselectivity when the aldehyde can chelate onto the metal center and form a rigid cyclic intermediate. The stereochemical outcome is then consistent with approach of the nucleophile anti to the more bulky substituent on the ring.[10][11]

Diels-Alder reaction
Lewis acids such as ZnCl2, BF3, SnCl4, AlCl3, and MeAlCl2 can catalyze both normal and inverse electron demand Diels-Alder reactions. The enhancement in rate is often dramatic, and regioselectivity towards ortho- or para-like products is often improved, as shown in the reaction between isoprene and methyl acrylate.[12]

The catalyzed Diels-Alder reaction is believed to be concerted. A computational study at the B3LYP/6-31G(d) level has shown, however, that the transition state of the BF3-catalyzed Diels-Alder reaction between propenal and 1,3-butadiene is more asynchronous than that of the thermal reaction – the bond further from the carbonyl group is formed ahead of the other bond.[13]
Ene reaction
The carbonyl-ene reaction is almost always catalyzed by Lewis acids in synthetic applications.[14] A stepwise or a largely asynchronous mechanism has been proposed for the catalyzed reaction based on kinetic isotope effect studies.[15] Nonetheless, cyclic transition states are frequently invoked to interpret diastereoselectivity. In a seminal review in the early 1990s, Mikami and colleagues[16] proposed a late, chair-like transition state, which could rationalize many observed stereochemical results, including the role of steric bulk in diastereoselectivity:[17]
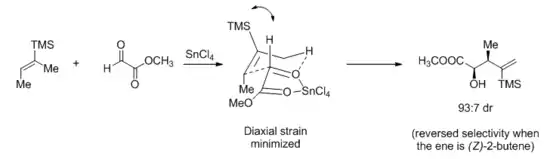
More recently, however, the same group carried out HF/6-31G* calculations on tin or aluminum Lewis acid-catalyzed ene reactions. Citing that methyl gloxylate chelates tin Lewis acids but not aluminum ones, they invoked an early, envelope-like transition state and rationalized the divergent stereochemical outcome of the ene reaction between (E)-2-butene and methyl glyoxylate.[18]

Application in synthesis
Lewis-acid catalyzed carbonyl addition reactions are routinely used to form carbon–carbon bonds in natural product synthesis. The first two reactions shown below are from the syntheses of (+)-lycoflexine[19] and zaragozic acid C,[20] respectively, which are direct applications of Sakurai and Mukaiyama reactions. The third reaction, en route to (+)-fawcettimine, is a Lewis-acid catalyzed cyclopropane opening that is analogous to a Mukaiyama-Michael reaction.[21]

The Diels-Alder reaction catalyzed or promoted by Lewis acids is a powerful and widely used method in natural product synthesis to attain scaffold complexity in a single step with stereochemical control. The two reaction shown below are an intramolecular Diels-Alder reaction towards (−)-fusarisetin A[22] and an intermolecular hetero-Diels-Alder reaction towards (−)-epibatidine,[23] respectively.

Friedel–Crafts and related reactions
In Friedel–Crafts alkylation, a Lewis acid – usually a simple metal halide salt – promotes heterolytic cleavage of a carbon–halogen bond in an alkyl halide and generates a carbocation, which undergoes electrophilic aromatic substitution. Although vastly useful in synthesis, the reaction often suffers from side reactions that arise from carbocation rearrangement, alkyl migration, and over-alkylation. Similarly, in Friedel-Crafts acylation, a Lewis acid assists in the generation of an acylium ion from an acid chloride (or occasionally acid anhydride). Although the acylium ion is often assumed to be the active intermediate,[24] there is evidence that the protonated acylium dication is the active electrophile that undergoes subsequent electrophilic aromatic substitution.[25]
Important variants of the Friedel–Crafts reaction include chloromethylation (with formaldehyde and HCl), formylation (with HCl and CO or CN−), and acylation with a nitrile as the acyl source. The nitrile-based acylation is particularly useful because it allows direct ortho-acylation of aniline without protecting the amine group.[26] A combination of a weak and a strong Lewis acid is necessary for the reaction to proceed, through the mechanism shown below. Guided by this mechanism, and equipped with knowledge that gallium trihalides are among the strongest Lewis acids,[27] process chemists at Merck were able to develop highly efficient conditions for this condition towards a drug candidate.[28]
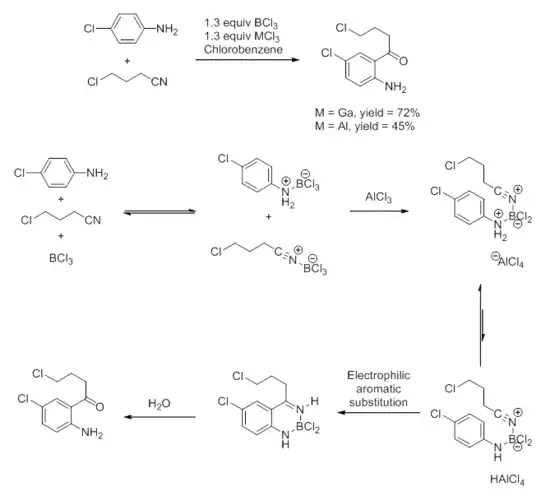
Asymmetric Lewis acid catalysis
Common Chiral Ligands
Asymmetric catalysis by Lewis acids rely on catalysts with chiral ligands coordinated to the metal center. Over the years, a small number of chiral ligand scaffolds have stood out as having "privileged" catalytic properties suitable for a wide range of applications, often of unrelated mechanisms. Current research efforts in asymmetric Lewis acid catalysis mostly utilize or modify those ligands rather than create new scaffolds de novo. The "privileged" scaffolds share a few common features, including chemical stability and relative ease of elaboration. The majority of the scaffolds are multidentate. Most of them also have high scaffold rigidity within the ligand. Several of them have fairly mature stereoinduction models available. Some "privileged" scaffolds, as identified by Jacobsen[29] and Zhou,[30] are introduced below.
Bisoxazolines (BOX)
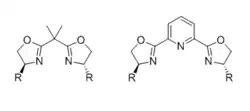
Most common chiral bisoxazoline (BOX) ligands consist of two identical chiral oxazoline moieties, substituted by a bulky group at the 4-positions, joined by a linker. The ligand is bidentate when the linker is a single carbon unit, but is tridentate (usually meridial) when the linker bears an additional coordinating atom, such as a pyridine nitrogen in the case of PyBOX ligands. The impact of ligand denticity and active intermediate geometry on the stereochemical outcome has been thoroughly reviewed.[31]
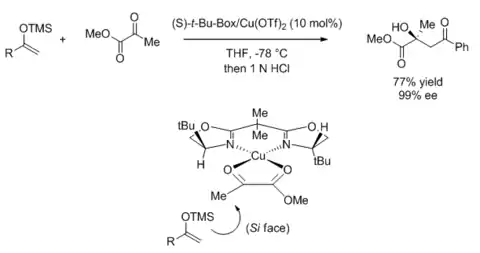
Many bidentate BOX-based Lewis acid-catalyzed reactions are based on copper(II) catalysts with substrates that are suitable for two-point binding. The stereochemical outcome is consistent with a twisted square planar intermediate that was proposed based on related crystal structures.[32][33] The substituent at the oxazoline's 4-position blocks one enantiotopic face of the substrate, leading to enantioselectivity. This is demonstrated in the following aldol-type reaction,[34] but is applicable to a wide variety of reactions such as Mannich-type reactions,[35] ene reaction,[36] Michael addition,[37] Nazarov cyclization,[38] and hetero-Diels-Alder reaction.[39]
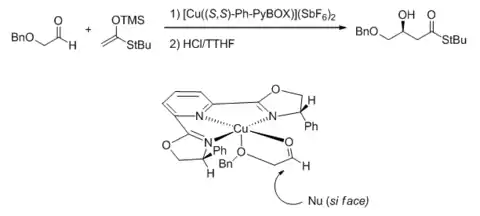
On the other hand, two-point binding on a Lewis acid bearing the meridially tridentate PyBOX ligand would result in a square pyramidal complex. A study using (benzyloxy)acetaldehyde as the electrophile showed that the stereochemical outcome is consistent with the carbonyl oxygen binding equatorially and the ether oxygen binding axially.[40]
BINAP
Developed by Noyori, BINAP (2,2'-diphenylphosphino-1,1'-binaphthyl) is a family of chiral diphosphine ligands featuring two triarylphosphine moieties installed on a binaphthalene backbone.[41] BINAP chelates onto a metal (usually a late transition metal) to form a C2-symmetric complex. As shown below in the structure of an (R)-BINAP ruthenium complex,[42] among the four remaining coordination sites on an octahedral metal center, the two equatorial sites (purple) are strongly influenced by the equatorial phenyl groups, while the two axial sites (green) are influenced by the axial phenyl groups.
-BINAP_Ligand_and_Complex.png.webp)
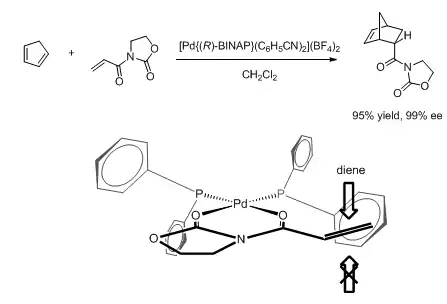
Based on the structure, models for the observed enantioselectivity in many BINAP-based Lewis acid-catalyzed reactions have been proposed. For example, in the palladium-catalyzed enantioselective Diels-Alder reaction shown below, the dienophile is thought to coordinate the metal center at the equatorial sites. Thus the equatorial phenyl group on phosphorus obstructs the Si-face, resulting in excellent enantioselectivity.[43] A very similar model was used to rationalize the outcome of a nickel-catalyzed asymmetric enolate alkylation reaction, where the substrate also bears an auxiliary that allows it to chelate onto the metal.[44] On the other hand, a copper(I)-catalyzed hetero-ene reaction is thought to proceed through a tetrahedral intermediate,[45] offering an alternative mode of stereoinduction by changing the metal center.
BINOL
BINOL (1,1'-binaphthyl-2,2'-diol) is usually used in conjunction with oxophilic Lewis acidic metals such as aluminum, titanium, zirconium, and various rare earth metals. In cases where BINOL itself does not provide ideal enantioselective control, it can be readily elaborated by substitution at the 3,3' positions (via lithiation) and 6,6' positions (via the 6,6'-dibromide compound prepared by electrophilic aromatic substitution) to modulate steric bulk and electronic properties.[46] For instance, aluminum catalysts based on bulky 3,3'-disilyl substituted BINOL have been developed as early examples of catalytic asymmetric hetero-Diels-Alder reaction[47] and Claisen rearrangement,[48] while introduction of electron-withdrawing groups at the 6,6'-positions was crucial for increasing the Lewis acidity, and hence catalytic activity, of zirconium(IV) catalysts toward a Mannich-type reaction.[49] To date, however, no model for the crucial factors governing BINOL-directed stereoinduction has been generally accepted.
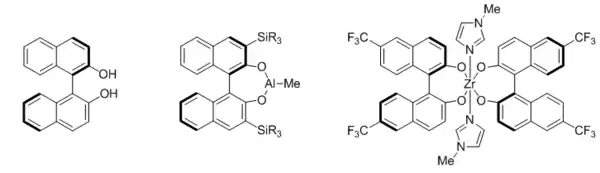
TADDOL
TADDOL stands for tetraaryl-1,3-dioxolane-4,5-dimethanol. The broad application of titanium TADDOLate catalysts towards carbonyl additions and cycloadditions has been introduced by Seebach and coworkers, and has been thoroughly summarized in a seminal review, in which a working stereoinduction model that agreed with the observed selectivity in a wide variety of reactions was put forth, despite the lack of a clear picture of the mechanism.[50]

Applications
Lewis acid catalysis has been used in the asymmetry-setting step for the syntheses of many natural products. The first reaction shown below, from the synthesis of taxane skeleton, uses a copper-based catalyst supported by a chiral phosphoramidite ligand for a conjugate carbonyl addition reaction.[51] The second reaction, from the synthesis of ent-hyperforin, uses an iron-PyBOX catalyst for an asymmetric Diels-Alder reaction.[52]

References
- Carey, Fransis A.; Sundberg, Richard J. (2007). Advanced Organic Chemistry : Part A: Structure and Mechanisms (5. ed.). Berlin: Springer US. ISBN 9780387683461.
- Yamamoto, Hisashi (ed.) (2000). Lewis acids in organic synthesis. Weinheim: Wiley-VCH. ISBN 978-3527295791.CS1 maint: extra text: authors list (link)
- Denmark, S.E.; Willson, T.M. in Selectivities in Lewis Acid Promoted Reactions, Schinzer, D., Ed.; Kluwer Academic Publishers, 1989, pp. 247–263.
- Yates, Peter; Eaton, Philip (20 August 1960). "Acceleration of the Diels-Alder Reaction by Aluminum Chloride". Journal of the American Chemical Society. 82 (16): 4436–4437. doi:10.1021/ja01501a085.
- Houk, K. N.; Strozier, R. W. (1 June 1973). "Lewis acid catalysis of Diels-Alder reactions". Journal of the American Chemical Society. 95 (12): 4094–4096. doi:10.1021/ja00793a070.
- Shambayati, S.; Schreiber, S.L. Lewis Acid Carbonyl Complexation, in Comprehensive Organic Synthesis, Trost, B.M.; Fleming, I., Eds. Pergamon, Oxford, 1991, vol. 1, chap. 1.10, pp. 283–324.
- Corcoran, Robert C.; Ma, Junning (1 June 1992). "Geometrical aspects of the activation of ketones by Lewis acids". Journal of the American Chemical Society. 114 (12): 4536–4542. doi:10.1021/ja00038a014.
- Hayashi, Tamio; Kabeta, Keiji; Hamachi, Itaru; Kumada, Makoto (1 January 1983). "Erythroselectivity in addition of γ-substituted allylsilanes to aldehydes in the presence of titanium chloride". Tetrahedron Letters. 24 (28): 2865–2868. doi:10.1016/S0040-4039(00)88045-4.
- Heathcock, Clayton H.; Hug, Kathleen T.; Flippin, Lee A. (1 January 1984). "Acyclic stereoselection. 27. Simple diastereoselection in the lewis acid mediated reactions of enolsilanes with aldehydes". Tetrahedron Letters. 25 (52): 5973–5976. doi:10.1016/S0040-4039(01)81736-6.
- Evans, David A.; Allison, Brett D.; Yang, Michael G.; Masse, Craig E. (1 November 2001). "The Exceptional Chelating Ability of Dimethylaluminum Chloride and Methylaluminum Dichloride. The Merged Stereochemical Impact of α- and β-Stereocenters in Chelate-Controlled Carbonyl Addition Reactions with Enolsilane and Hydride Nucleophiles". Journal of the American Chemical Society. 123 (44): 10840–10852. doi:10.1021/ja011337j. PMID 11686685.
- Heathcock, Clayton H.; Kiyooka, Syunichi; Blumenkopf, Todd A. (1 November 1984). "Acyclic stereoselection. 22. Diastereofacial selectivity in the Lewis acid mediated reactions of allylsilanes with chiral aldehydes and enones". The Journal of Organic Chemistry. 49 (22): 4214–4223. doi:10.1021/jo00196a022.
- Inukai, Takashi; Kojima, Takeshi (1 April 1966). "Catalytic Actions of Aluminum Chloride on the Isoprene—Methyl Acrylate Diels-Alder Reaction". The Journal of Organic Chemistry. 31 (4): 1121–1123. doi:10.1021/jo01342a031.
- García, J. I.; Martínez-Merino, V.; Mayoral, J. A.; Salvatella, L. (1 March 1998). "Density Functional Theory Study of a Lewis Acid Catalyzed Diels−Alder Reaction. The Butadiene + Acrolein Paradigm". Journal of the American Chemical Society. 120 (10): 2415–2420. doi:10.1021/ja9722279.
- Carey, Francis A.; Sundberg, Richard J. (2007). Reactions and synthesis (5. ed.). New York, NY: Springer. p. 871. ISBN 9780387683508.
- Singleton, Daniel A.; Hang, Chao (1 February 2000). "13C and 2H Kinetic Isotope Effects and the Mechanism of Lewis Acid-Catalyzed Ene Reactions of Formaldehyde". The Journal of Organic Chemistry. 65 (3): 895–899. doi:10.1021/jo9917590. PMID 10814025.
- Mikami, Koichi; Shimizu, Masaki (1 July 1992). "Asymmetric ene reactions in organic synthesis". Chemical Reviews. 92 (5): 1021–1050. doi:10.1021/cr00013a014.
- Mikami, Koichi; Loh, Teck Peng; Nakai, Takeshi (1 August 1990). "Carbonyl-ene reaction with vinylsilanes: silicon as a controlling element for regio- and stereochemistry". Journal of the American Chemical Society. 112 (18): 6737–6738. doi:10.1021/ja00174a058.
- Yamanaka, Masahiro; Mikami, Koichi (1 December 2002). "Theoretical Studies on the Diastereoselectivity in the Lewis Acid Catalyzed Carbonyl-Ene Reaction: A Fundamental Role of Electrostatic Interaction". Helvetica Chimica Acta. 85 (12): 4264–4271. doi:10.1002/hlca.200290011.
- Ramharter, Jürgen; Weinstabl, Harald; Mulzer, Johann (20 October 2010). "Synthesis of the Lycopodium Alkaloid (+)-Lycoflexine". Journal of the American Chemical Society. 132 (41): 14338–14339. doi:10.1021/ja107533m. PMID 20866095.
- Evans, David A.; Barrow, James C.; Leighton, James L.; Robichaud, Albert J.; Sefkow, Michael (1 December 1994). "Asymmetric Synthesis of the Squalene Synthase Inhibitor Zaragozic Acid C". Journal of the American Chemical Society. 116 (26): 12111–12112. doi:10.1021/ja00105a085.
- Jung, Michael E.; Chang, Jonah J. (2 July 2010). "Enantiospecific Formal Total Synthesis of (+)-Fawcettimine". Organic Letters. 12 (13): 2962–2965. doi:10.1021/ol1009762. PMID 20515058.
- Deng, Jun; Zhu, Bo; Lu, Zhaoyong; Yu, Haixin; Li, Ang (18 January 2012). "Total Synthesis of (−)-Fusarisetin A and Reassignment of the Absolute Configuration of Its Natural Counterpart". Journal of the American Chemical Society. 134 (2): 920–923. doi:10.1021/ja211444m. PMID 22239597.
- Evans, David A.; Scheidt, Karl A.; Downey, C. Wade (1 September 2001). "Synthesis of (−)-Epibatidine". Organic Letters. 3 (19): 3009–3012. doi:10.1021/ol016420q. PMID 11554830.
- Olah, G. A. (ed.) Friedel-Crafts and Related Reactions Interscience, New York, 1964
- Sato, Yasuo; Yato, Michihisa; Ohwada, Tomohiko; Saito, Shinichi; Shudo, Koichi (1 March 1995). "Involvement of Dicationic Species as the Reactive Intermediates in Gattermann, Houben-Hoesch, and Friedel-Crafts Reactions of Nonactivated Benzenes". Journal of the American Chemical Society. 117 (11): 3037–3043. doi:10.1021/ja00116a009.
- Sugasawa, Tsutomu; Toyoda, Tatsuo; Adachi, Makoto; Sasakura, Kazuyuki (1 July 1978). "Aminohaloborane in organic synthesis. 1. Specific ortho substitution reaction of anilines". Journal of the American Chemical Society. 100 (15): 4842–4852. doi:10.1021/ja00483a034.
- Olah, George A.; Kobayashi, Shiro; Tashiro, Masashi (1 October 1972). "Aromatic substitution. XXX. Friedel-Crafts benzylation of benzene and toluene with benzyl and substituted benzyl halides". Journal of the American Chemical Society. 94 (21): 7448–7461. doi:10.1021/ja00776a030.
- Yasuda, Nobuyoshi (ed.) (2009). The Art of Process Chemistry. Weinheim, Bergstr: Wiley-VCH. pp. 1–43. ISBN 9783527324705.CS1 maint: extra text: authors list (link)
- Yoon, T. P.; Jacobsen, Eric N. (14 March 2003). "Privileged Chiral Catalysts". Science. 299 (5613): 1691–1693. Bibcode:2003Sci...299.1691Y. doi:10.1126/science.1083622. PMID 12637734.
- Zhou, Qi-Lin (ed.) (2011). Privileged chiral ligands and catalysts. Weinheim, Germany: Wiley-VCH. ISBN 9783527327041.CS1 maint: extra text: authors list (link)
- Rasappan, Ramesh; Laventine, Dominic; Reiser, Oliver (1 March 2008). "Metal-bis(oxazoline) complexes: From coordination chemistry to asymmetric catalysis". Coordination Chemistry Reviews. 252 (5–7): 702–714. doi:10.1016/j.ccr.2007.11.007.
- Evans, David A.; Miller, Scott J.; Lectka, Thomas; von Matt, Peter (1 August 1999). "Chiral Bis(oxazoline)copper(II) Complexes as Lewis Acid Catalysts for the Enantioselective Diels−Alder Reaction". Journal of the American Chemical Society. 121 (33): 7559–7573. doi:10.1021/ja991190k.
- Thorhauge, Jacob; Roberson, Mark; Hazell, Rita G.; Jørgensen, Karl Anker (15 April 2002). "On the Intermediates in Chiral Bis(oxazoline)copper(II)-Catalyzed Enantioselective Reactions—Experimental and Theoretical Investigations". Chemistry: A European Journal. 8 (8): 1888. doi:10.1002/1521-3765(20020415)8:8<1888::AID-CHEM1888>3.0.CO;2-9.
- Evans, David A.; Burgey, Christopher S.; Kozlowski, Marisa C.; Tregay, Steven W. (1 February 1999). "-Symmetric Copper(II) Complexes as Chiral Lewis Acids. Scope and Mechanism of the Catalytic Enantioselective Aldol Additions of Enolsilanes to Pyruvate Esters". Journal of the American Chemical Society. 121 (4): 686–699. doi:10.1021/ja982983u.
- Marigo, Mauro; Kjærsgaard, Anne; Juhl, Karsten; Gathergood, Nicholas; Jørgensen, Karl Anker (23 May 2003). "Direct Catalytic Asymmetric Mannich Reactions of Malonates and -Keto Esters". Chemistry: A European Journal. 9 (10): 2359–2367. doi:10.1002/chem.200204679. PMID 12772311.
- Evans, David A.; Burgey, Christopher S.; Paras, Nick A.; Vojkovsky, Tomas; Tregay, Steven W. (1 June 1998). "C2-Symmetric Copper(II) Complexes as Chiral Lewis Acids. Enantioselective Catalysis of the Glyoxylate−Ene Reaction". Journal of the American Chemical Society. 120 (23): 5824–5825. doi:10.1021/ja980549m.
- Evans, David A.; Willis, Michael C.; Johnston, Jeffrey N. (1 September 1999). "Catalytic Enantioselective Michael Additions to Unsaturated Ester Derivatives Using Chiral Copper(II) Lewis Acid Complexes". Organic Letters. 1 (6): 865–868. doi:10.1021/ol9901570. PMID 10823215.
- Aggarwal, Varinder K.; Belfield, Andrew J. (1 December 2003). "Catalytic Asymmetric Nazarov Reactions Promoted by Chiral Lewis Acid Complexes". Organic Letters. 5 (26): 5075–5078. doi:10.1021/ol036133h. PMID 14682768.
- Yao, Sulan; Johannsen, Mogens; Audrain, Hélène; Hazell, Rita G.; Jørgensen, Karl Anker (1 September 1998). "Catalytic Asymmetric Hetero-Diels−Alder Reactions of Ketones: Chemzymatic Reactions". Journal of the American Chemical Society. 120 (34): 8599–8605. doi:10.1021/ja981710w.
- Evans, David A.; Kozlowski, Marisa C.; Murry, Jerry A.; Burgey, Christopher S.; Campos, Kevin R.; Connell, Brian T.; Staples, Richard J. (1 February 1999). "C2-Symmetric Copper(II) Complexes as Chiral Lewis Acids. Scope and Mechanism of Catalytic Enantioselective Aldol Additions of Enolsilanes to (Benzyloxy)acetaldehyde". Journal of the American Chemical Society. 121 (4): 669–685. doi:10.1021/ja9829822.
- Miyashita, A.; Takaya, H.; Souchi, T.; Noyori, R. (1 January 1984). "2, 2'-bis(diphenylphosphino)-1, 1'-binaphthyl(binap)". Tetrahedron. 40 (8): 1245–1253. doi:10.1016/S0040-4020(01)82411-X.
- Akotsi, Okwado M.; Metera, K; Reid, R.D.; McDonald, R; Bergens, S. H. (19 May 2000). "Versatile precursor to ruthenium-bis(phosphine) hydrogenation catalysts". Chirality. 12 (5–6): 514–522. doi:10.1002/(SICI)1520-636X(2000)12:5/6<514::AID-CHIR38>3.0.CO;2-#.
- Ghosh, Arun K.; Matsuda, Hideho (1 December 1999). "Counterions of BINAP−Pt(II) and −Pd(II) Complexes: Novel Catalysts for Highly Enantioselective Diels−Alder Reaction". Organic Letters. 1 (13): 2157–2159. doi:10.1021/ol990346i. PMID 10836069.
- Evans, David A.; Thomson, Regan J. (1 August 2005). "Ni(II) Tol-BINAP-Catalyzed Enantioselective Orthoester Alkylations of N-Acylthiazolidinethiones". Journal of the American Chemical Society. 127 (30): 10506–10507. doi:10.1021/ja053386s. PMID 16045335.
- Yamamoto, Yuhei; Yamamoto, Hisashi (1 April 2004). "Catalytic, Highly Enantio, and Diastereoselective Nitroso Diels−Alder Reaction". Journal of the American Chemical Society. 126 (13): 4128–4129. doi:10.1021/ja049849w. PMID 15053601.
- Chen, Yu; Yekta, Shahla; Yudin, Andrei K. (1 August 2003). "Modified BINOL Ligands in Asymmetric Catalysis". Chemical Reviews. 103 (8): 3155–3212. doi:10.1021/cr020025b. PMID 12914495.
- Maruoka, Keiji.; Itoh, Takayuki.; Shirasaka, Tadashi.; Yamamoto, Hisashi. (1 January 1988). "Asymmetric hetero-Diels-Alder reaction catalyzed by a chiral organoaluminum reagent". Journal of the American Chemical Society. 110 (1): 310–312. doi:10.1021/ja00209a061.
- Maruoka, Keiji; Hiroshi Banno; Hisashi Yamamoto (1990). "Asymmetric Claisen rearrangement catalyzed by chiral organoaluminum reagent". J. Am. Chem. Soc. 112 (21): 7791–7793. doi:10.1021/ja00177a047.
- Ishitani, Haruro; Ueno, Masaharu; Kobayashi, Shū (1 August 2000). "Enantioselective Mannich-Type Reactions Using a Novel Chiral Zirconium Catalyst for the Synthesis of Optically Active β-Amino Acid Derivatives". Journal of the American Chemical Society. 122 (34): 8180–8186. doi:10.1021/ja001642p.
- Seebach, Dieter; Beck, Albert K.; Heckel, Alexander (5 January 2001). "TADDOLs, Their Derivatives, and TADDOL Analogues: Versatile Chiral Auxiliaries". Angewandte Chemie International Edition. 40 (1): 92–138. doi:10.1002/1521-3773(20010105)40:1<92::AID-ANIE92>3.0.CO;2-K.
- Mendoza, Abraham; Ishihara, Yoshihiro; Baran, Phil S. (6 November 2011). "Scalable enantioselective total synthesis of taxanes". Nature Chemistry. 4 (1): 21–25. Bibcode:2012NatCh...4...21M. doi:10.1038/nchem.1196. PMC 3243931. PMID 22169867.
- Shimizu, Yohei; Shi, Shi-Liang; Usuda, Hiroyuki; Kanai, Motomu; Shibasaki, Masakatsu (1 February 2010). "Catalytic Asymmetric Total Synthesis of ent-Hyperforin". Angewandte Chemie International Edition. 49 (6): 1103–1106. doi:10.1002/anie.200906678. PMID 20063336.