LPS-responsive beige-like anchor protein deficiency
LPS-responsive beige-like anchor protein deficiency is a rare genetic condition caused by the absence of LPS-responsive beige-like anchor protein (LRBA).
| LPS-responsive beige-like anchor protein deficiency | |
|---|---|
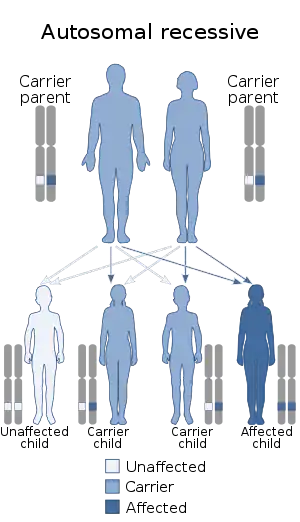 | |
| This condition is inherited in an autosomal recessive manner |
Signs and symptoms
The presentation of this condition is variable making the diagnosis difficult. The most common features include[1]
- Immune dysregulation (95%)
- Organomegaly (86%)
- Recurrent infections (71%)
- Hypogammaglobulinemia (57%)
- Granulomatous lymphocytic interstitial lung disease (38%)
There is also a tendency to develop inflammatory bowel disease.
Genetics
The LBRA gene is located on the long arm of chromosome 4 (4q31.3).
Pathogenesis
LBRA protein interacts with the protein CTLA4. The absence of LBRA increases the turnover of CTLA4 and interferes with vesicle trafficing.
Diagnosis
Differential diagnosis
Management
Along with treatment for infections and other complications several additional treatments have been tried. These include hematopoietic stem cell transplantation, immunoglobulin replacement and immunosuppressive treatment.[1]
History
This condition was first described in 2012.[2]
References
- Gámez-Díaz L, August D, Stepensky P, Revel-Vilk S, Seidel MG, Noriko M, Morio T, Worth AJJ, Blessing J, Van de Veerdonk F, Feuchtinger T, Kanariou M, Schmitt-Graeff A, Jung S, Seneviratne S, Burns S, Belohradsky BH, Rezaei N, Bakhtiar S, Speckmann C, Jordan M, Grimbacher B (2016) The extended phenotype of LPS-responsive beige-like anchor protein (LRBA) deficiency. J Allergy Clin Immunol 137(1):223-230
- Lopez-Herrera G, Tampella G, Pan-Hammarstrom Q, Herholz P, Trujillo-Vargas CM, Phadwal K, Simon AK, Moutschen M, Etzioni A, Mory A, Srugo I, Melamed, D and 21 others. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet 90: 986-1001