Intramolecular reactions of diazocarbonyl compounds
Intramolecular reactions of diazocarbonyl compounds include addition to carbon–carbon double bonds to form fused cyclopropanes and insertion into carbon–hydrogen bonds or carbon–carbon bonds.[1]
Introduction
In the presence of an appropriate transition metal (typically copper or rhodium [2]), α-diazocarbonyl compounds are converted to transition metal carbenes, which undergo addition reactions in the presence of carbon–carbon double bonds to form cyclopropanes.[3] Insertion into carbon–carbon or carbon–hydrogen bonds is possible in substrates lacking a double bond.[4] The intramolecular version of this reaction forms fused carbocycles, although yields of reactions mediated by copper are typically moderate. For enantioselective cyclopropanations and insertions, both copper- and rhodium-based catalysts are employed, although the latter have been more heavily studied in recent years.[5]
(1)
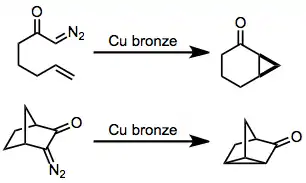
Mechanism and stereochemistry
Prevailing mechanism
The reaction mechanism of decomposition of diazocarbonyl compounds with copper begins with the formation of a copper carbene complex. Evidence for the formation of copper carbenes is provided by comparison to the behavior of photolytically generated free carbenes[6] and the observation of appreciable enantioselectivity in cyclopropanations with chiral copper complexes.[7] Upon formation of the copper carbene, either insertion or addition takes place to afford carbocycles or cyclopropanes, respectively. Both addition and insertion proceed with retention of configuration.[8][9] Thus, diastereoselectivity may often be dictated by the configuration of the starting material.
(2)
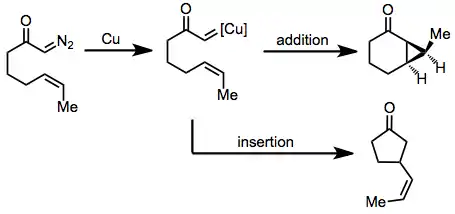
Scope and limitations
Either copper powder or copper salts can be used very generally for intramolecular reactions of diazocarbonyl compounds. This section describes the different types of diazocarbonyl compounds that may undergo intramolecular reactions in the presence of copper. Note that for intermolecular reactions of diazocarbonyl compounds, the use of rhodium catalysts is preferred.[2]
Diazoketones containing pendant double bonds undergo cyclopropanation in the presence of copper. The key step in one synthesis of barbaralone is the selective intramolecular cyclopropanation of a cycloheptatriene.[10]
(3)
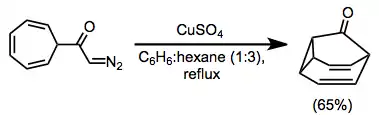
α,β-Cyclopropyl ketones may act as masked α,β-unsaturated ketones. In one example, intramolecular participation of an aryl group leads to the formation of a polycyclic ring system with complete diastereoselectivity.[11]
(4)

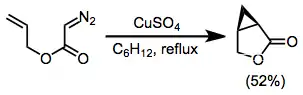
α-Diazoesters are not as efficient as diazoketones at intramolecular cyclizations in some cases because of the propensity of esters to exist in the trans conformation about the carbon–oxygen single bond.[12] However, intramolecular reactions of diazoesters do take place—in the example in equation (5), copper(II) sulfate is used to effect the formation of the cyclopropyl ester shown.[6]
(5)
In the presence of a catalytic amount of acid, diazomethyl ketone substrates containing a pendant double bond or aryl group undergo cyclization. The mechanism of this process most likely involves protonation of the diazocarbonyl group to form a diazonium salt, followed by displacement of nitrogen by the unsaturated functionality and deprotonation. In the example below, demethylation affords a quinone.[13]
(6)

When no unsaturated functionality is present in the substrate, C-H insertion may occur. C-H Insertion is particularly facile in conformationally restricted substrates in which a C-H bond is held in close proximity to the diazo group.[14]
(7)
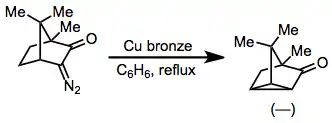
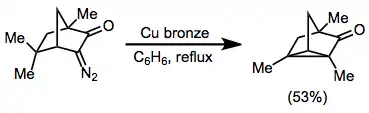
Transannular insertions, which form fused carbocyclic products, have also been observed. Yields are often low for these reactions, however.[15]
(8)
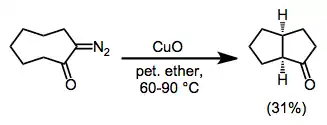
Insertion into carbon–carbon bonds has been observed. In the example in equation (9), the methyl group is held in close proximity to the diazo group, facilitating C-C insertion.[14]
(9)
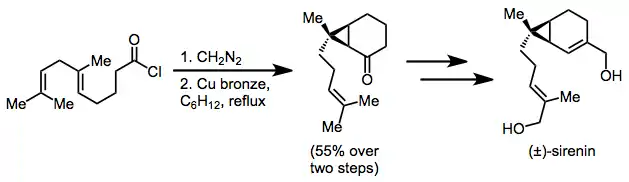
Synthetic applications
Intramolecular cyclopropanation of a diazoketone is applied in a racemic synthesis of sirenin. A single cyclopropane diastereomer was isolated in 55% yield after diazoketone formation and cyclization.[16]
(10)
Experimental conditions and procedure
Typical conditions
Diazo compounds may be explosive and should be handled with care. Very often, the diazocarbonyl compound is prepared and immediately used via treatment of the corresponding acid chloride with an excess of diazomethane (see Eq. (18) below for an example).[17] Reactions mediated by copper are typically on the order of hours, and in some cases, slow addition of the diazocarbonyl compound is necessary. Reactions should be carried out under an inert atmosphere in anhydrous conditions.
Example procedure[18]
(11)
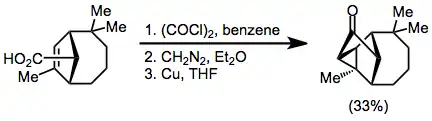
A solution of the olefinic acid (0.499 g, 2.25 mmol) dissolved in benzene (20 ml, freshly distilled from calcium hydride) was stirred at 0 °C (ice bath) under nitrogen while oxalyl chloride (1.35 ml, 2.0 g, 15.75 mmol) was added dropwise. The ice bath was removed and the solution was stirred at room temperature for 2 hr. The solvent and excess reagent were removed in vacuo. The resulting orange oil was dissolved in benzene (2 x 5.0 mi, freshly distilled from calcium hydride) under nitrogen.
This solution was added dropwise at 0 °C (ice bath) to an anhydrous ethereal solution of diazomethane (50 ml, −20 mmol, predried over sodium metal) with vigorous stirring under nitrogen. The resulting solution was stirred at 0 °C for 1 hr and then at room temperature for 1.5 hr. The solvents and excess reagent were removed in vacuo.
Tetrahydrofuran (40 ml, freshly distilled from lithium aluminum hydride) and finely divided metallic copper powder (0.67 g) were added to the crude diazo ketone, sequentially. This suspension was vigorously stirred at reflux under nitrogen for 2 hr. The resulting suspension was allowed to stir at room temperature for an additional 14 hr. The solution was filtered into water (100 ml). The mixture was shaken vigorously for 5 min and then extracted with ether (3 x 50 ml). The combined ethereal extracts were washed with saturated sodium bicarbonate solution (4 X 40 ml), water (40 ml), and saturated sodium chloride solution (40 ml), dried (Na2SO4), and concentrated in vacuo to give 0.673 g of a crude brown oil. This crude oil was chromatographed on silica gel (67 g) in a 2-cm diameter column using 10% ether-90% petroleum ether to develop the column, taking 37-ml sized fractions. Fractions 11–16 gave 0.164 g (33%) of pure ketone product: mp 64-64.5° (from pentane); IR (CCl4) 3095 (cyclopropyl CH) and 1755 cm−1 (CO); NMR (CCl4) δ 1.18 (s, 3H, CH3) 1.03 (9, 3H, CH3), 0.97 (s, 3H, CH3), and 0.90 ppm (s, 3H, CH3). Anal. Calcd for C15H22O: C, 82.52; H, 10.16. Found: C, 82.61; H, 10.01.
References
- Burke, S. D.; Grieco, P. A. Org. React. 1979, 26, 361. doi:10.1002/0471264180.or026.02
- Davies, H.; Antoulinakis, E. Org. React. 2004, 57, 1.
- Stork, G.; Ficini, J. J. Am. Chem. Soc. 1961, 83, 4678.
- Nakata, T.; Tahara, A. Tetrahedron Lett. 1976, 1515.
- Doyle, M.; Forbes, D. Chem. Rev. 1998, 98, 911.
- Kirmse, W.; Dietrich, H. Chem. Ber., 1965, 98, 4027.
- Fritschi, H.; Leutenegger, U. Angew. Chem. Int. Ed. 2000, 25, 1005.
- Stork, G.; Gregson, M. J. Am. Chem. Soc. 1969, 91, 2372, footnote 6.
- Ledon, H.; Linstrumelle, G.; Julia, S. Tetrahedron Lett. 1973, 25.
- Doering, W.; Ferrier, B.; Fossel, E.; Hartenstein, J.; Jones, Jr., M.; Klumpp, G.; Rubin, R.; Saunders, M. Tetrahedron 1967, 23, 3943.
- Stork, G.; Gregson, M.; J. Am. Chem. Soc. 1969, 91, 2373.
- Rando, R.; J. Am. Chem. Soc. 1970, 92, 6706.
- Beames, D.; Klose, T.; Mander, L. Aust. J. Chem. 1974, 27, 1269.
- Yates, P.; Danishefsky, S. J. Am. Chem. Soc. '1962, 84, 879.
- Regitz, M.; Rüter, J. Chem. Ber. 1969, 102, 3877.
- Grieco, P. J. Am. Chem. Soc. 1969, 91, 5660.
- House, H.; Boots, S.; Jones, V. J. Org. Chem. 1965, 30, 2519.
- Welch, S. C.; Walters, R. L. J. Org. Chem. 1974, 39, 2665.