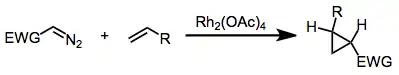
Intermolecular metal-catalyzed carbenoid cyclopropanations
Metal-catalyzed intermolecular carbenoid cyclopropanations are organic reactions that result in the formation of a cyclopropane ring from a metal carbenoid specie and an alkene.[1] In the Simmons–Smith reaction the metal involved is zinc.
Introduction
Cyclopropanes may be formed through the reaction of a metal carbenoid specie (generated through the reaction of a diazo compound with a transition metal) and an olefin. Although the intramolecular variant of this reaction has been known since 1961,[2] chemo- and stereoselective intermolecular metal-catalyzed cyclopropanation reactions employing diazocarbonyl compounds are more recent. Rhodium carboxylate complexes, such as dirhodium tetraacetate, are most commonly used to catalyze this transformation. Enantioselective cyclopropanations have been developed and typically make use of pre-formed chiral rhodium carboxylate complexes derived from chiral carboxylate ligands.[3]
(1)
Mechanism and Stereochemistry
Prevailing Mechanism
Definitive mechanistic studies of rhodium-catalyzed cyclopropanation are lacking. However, the mechanism has been rationalized based on product distribution and stereoselectivity.[4] Attack of the diazo compound on the metal center generates a zwitterionic metal alkyl complex, which expels nitrogen gas to afford a metal carbene intermediate. Concerted addition of the metal carbene to the olefin (without direct coordination of the olefin to the metal) generates the observed cyclopropane product.[5] The configuration of the olefin is retained throughout the process;[6] however, metal carbenes with heterotopic faces may generate a mixture of diastereomers, as shown at the right of Eq. (2).
(2)
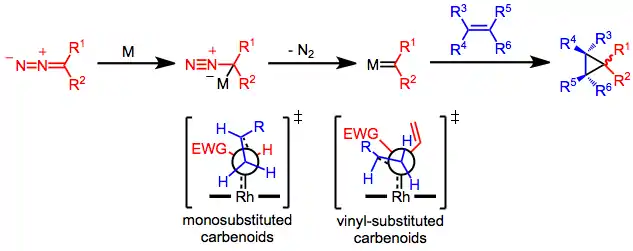
The configuration of the product is determined by the trajectory of approach of the olefin to the metal carbene. In reactions of monosubstituted metal carbenes with terminal olefins, the olefin likely approaches "end-on" (with the carbon-carbon double bond of the olefin nearly parallel to the metal-carbon double bond of the carbene) with the olefin R group pointed away from the substituent of the carbene.[7] A second transition state model has been proposed for reactions of vinyl-substituted carbenes. In this model, the olefin approaches "side-on" (with the carbon-carbon double bond of the olefin perpendicular to the metal-carbon double bond of the carbene) with the olefin R group far from the vinyl group.[8]
Stereoselective Variants
Methods for the stereoselective synthesis of cyclopropanes from diazocarbonyl compounds and olefins have relied on either the use of pre-formed chiral rhodium catalysts or chiral auxiliaries on the diazocarbonyl compound. For example, Rh2[S-DOSP]4 is a highly effective catalyst for the enantioselective cyclopropanation of alkenes.[9]
(3)

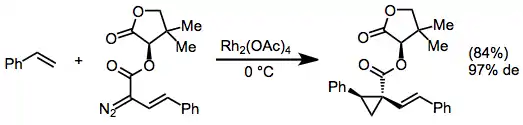
Chiral auxiliaries derived from readily available chiral alcohols (such as pantolactone) may be used for diastereoselective cyclopropanations with diazo esters.[10]
(4)
Scope and Limitations
Cyclopropanation of olefins with diazocarbonyl compounds is most commonly accomplished using rhodium carboxylate complexes, although copper was originally used.[11] The scope of the olefin is generally quite broad—electron-rich,[12] neutral,[13] and electron-poor[14] olefins have all been cyclopropanated efficiently using rhodium-based catalyst systems. This section describes the various classes of diazocarbonyl compounds that react with olefins under rhodium catalysis to afford cyclopropanes.
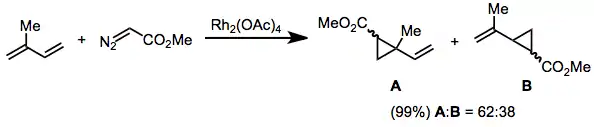
Diazoacetates that possess a single carbonyl substituent attached to the diazo carbon, have been used for the cyclopropanation of a wide array of olefins. Diastereoselectivity for the (E) cyclopropane increases as the size of the ester group increases. In addition, adding electron density to the catalyst (for instance by replacing acetate ligands with acetamide, acam) increases the diastereoselectivity of the reaction.[15]
(5)

Diazocarbonyl compounds substituted with two electron-withdrawing groups, such as diazomalonates, are prone to experience side reactions under cyclopropanation conditions. [3+2] Cycloaddition[16] and C-H insertion[17] side products have been observed.
(6)

Diazoacetates substituted with a vinyl or aryl group on the diazo carbon are unreactive towards trans-alkenes. This result has been explained by invoking the transition state model in Eq. (2). Reactions of these substrates are highly selective for the (E) cyclopropane isomer.[18]
(7)

Vinyl diazoacetates react with dienes to afford divinyl cyclopropanes, which undergo Cope rearrangement to afford cycloheptadienes.[19] The more substituted double bond of the diene reacts preferentially.[20]
(8)
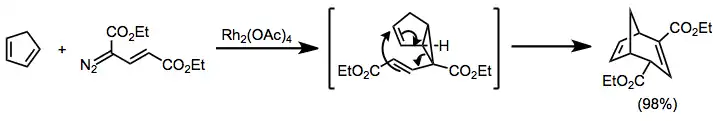
(9)
Furans react similarly with vinyl diazoacetates, although the intermediate cyclopropane may transform either into the Cope rearrangement product or an opened unsaturated carbonyl compound. The distribution of these products is highly dependent on the substitution pattern of the furan.[21]
(10)

Pyrroles react with vinyl diazoacetates to form nitrogen-bridged cycloheptadienes. The use of methyl lactate as a chiral auxiliary on the vinyl diazoacetate led to moderate diastereoselectivity in the tandem cyclopropanation/Cope rearrangement of Boc-protected pyrrole.[22]
(11)

Synthetic Applications
Enantioselective intermolecular cyclopropanation has been applied to the synthesis of chiral cyclopropane antibiotics, such as cilastatin.[23] (12) Tandem cyclopropanation/fragmentation is a key step in the synthesis of 12-hydroxyeicosatetraenoic acid.[24]
(12)

Comparison with Other Methods
Simmons-Smith cyclopropanation, which employs carbenes derived from diethylzinc and diiodomethane, is a popular alternative to rhodium-catalyzed cyclopropanation. In the presence of a chiral diamine, Simmons-Smith cyclopropanation is enantioselective; however, selectivities are not as high as the corresponding rhodium-catalyzed reactions.[25]
(13)

Substituted zinc carbenoids can be prepared from the corresponding ketones or aldehydes through a sequence analogous to the mechanism of the Clemmensen reduction. Cyclopropanation of olefins with these intermediates occurs with moderate diastereoselectivity and yield.[26]
(14)

Other diazo compounds besides diazocarbonyl compounds have been used for rhodium-catalyzed cyclopropanations;[27] however, these substrates are much more difficult to handle and unstable than diazocarbonyl compounds. Thus, they have not been extensively adopted for organic synthesis.
(15)

Experimental Conditions and Procedure
Typical Conditions
Great care should be taken in handling diazo compounds, which are toxic and potentially explosive. Reactions should be carried out in a well-ventilated fume hood behind a blast shield.
Rhodium(II) carboxylate complexes are easily prepared and indefinitely stable in air. Carbene dimerization is a significant problem in these reactions, but can be overcome through slow addition of the diazo compound or use of a large excess of alkene. The reaction is usually carried out under inert atmosphere in anhydrous conditions, and the most common solvent used is dichloromethane. However, the enantioselectivity of asymmetric cyclopropanations may depend profoundly on the solvent.[28]
Example Procedure[9]
(16)
A mixture of styrene (44.2 g, 424 mmol) and Rh2(S-DOSP)4 (1.58 g, 0.85 mmol) in pentane (350 mL) was stirred at −78° under an argon atmosphere. To this solution was added methyl (E)-2-diazo-4-phenyl-3-butenoate (17.2 g, 84.8 mmol) in pentane (0.12 M) over 30 minutes, and the reaction mixture was then stirred at −78° for 24 hours. The mixture was then concentrated in vacuo, and the residue was purified on silica using ether/petroleum ether (0:100 to 10:90) as the eluent to give (1S,2S)-methyl 2β-phenyl-1β-[2-(Z)-styryl]cyclopropane-1α-carboxylate (16.05 g, 68%) as a white solid (mp 57–60°; 98% ee); IR ( CHCl3) 3110, 3090, 3060, 2980, 2950, 2880, 1735 cm–1; 1H NMR (148) ( CDCl3) 1.85 (dd, J = 7.3, 5.1 Hz, 1 H), 2.05 (dd, J = 9.1, 5.1 Hz, 1 H), 3.04 (dd, J = 9.1, 7.3 Hz, 1 H), 3.77 (s, 3 H), 6.15 (d, J = 15.9, 1 H), 6.37, (d, J = 15.9 Hz, 1 H), 7.12–7.28 (m, 10 H); 13C NMR ( CDCl3) 18.5, 33.2, 34.9, 52.3, 124.0, 126.1, 126.7, 127.2, 127.9, 128.3, 129.0, 133.0, 135.4, 137.0, 174.1; [α]25D – 166° (c 1.1, CHCl3); Anal. Calcd. for C19H18O2: C, 81.99; H, 6.52. Found: C, 81.74; H, 6.53.
References
- Davies, H. M. L.; Antoulinakis, E. G. Org. React. 2001, 57, 1. doi:10.1002/0471264180.or057.01
- Burke, S. D.; Grieco, P. A. Org. React. 1979, 26, 361.
- Singh, V. K.; Arpita, D.; Sekar, G. Synthesis 1997, 137.
- Doyle, M. P. Acc. Chem. Res. 1986, 19, 348.
- Doyle, M. P.; McKervey, M. A.; Ye, T. In Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides; Wiley: New York, 1998, pp. 163-220.
- Doyle, M. P. Chem. Rev. 1986, 86, 919.
- Doyle, M. P.; Griffin, J. H.; Bagheri, V.; Dorow, R. L. Organometallics 1984, 3, 53.
- Davies, H. M. L. Curr. Org. Chem. 1998, 2, 463.
- Davies, H. M. L.; Bruzinski, P. R.; Lake, D. H.; Kong, N.; Fall, M. J. J. Am. Chem. Soc. 1996, 118, 6897.
- Davies, H. M. L.; Huby, N. J. S.; Cantrell, W. R., Jr.; Olive, J. L. J. Am. Chem. Soc. 1993, 115, 9468.
- Dave, V.; Warnhoff, E. Org. React. 1970, 18, 217.
- Ye, T.; McKervey, M. Chem. Rev. 1994, 94, 1091.
- Maas, G. Top. Curr. Chem. 1987, 137, 75.
- Doyle, M.; Dorow, R.; Buhro, W.; Griffin, J.; Tamblyn, W.; Trudell, M. Organometallics 1984, 3, 44.
- Doyle, M.; Bagheri, V.; Wandless, T.; Harn, N. K.; Brinker, D. A.; Eagle, C.; Loh, K. J. Am. Chem. Soc. 1990, 112, 1906.
- Pirrung, M. C.; Zhang, J.; Lackey, K.; Sternbach, D. D.; Brown, F. J. Org. Chem. 1995, 60, 2112.
- Peace, B. W.; Wulfman, D. S. Synthesis 1973, 137.
- Davies, H. M. L.; Clark, T. J.; Church, L. A. Tetrahedron Lett. 1989, 30, 5057.
- Davies, H.; Smith, H.; Korkor, O. Tetrahedron Lett. 1987, 28, 1853.
- Doyle, M.; Dorow, R.; Tamblyn, W.; Buhro, W. Tetrahedron Lett. 1982, 23, 2261.
- Wenkert, E. In New Trends in Natural Products Chemistry, Studies in Organic Chemistry; Rahman, A., Quesne, P. W., Eds.; Elsevier: Amsterdam, 1986; Vol. 26, pp. 557–563
- Davies, H. M. L.; Huby, N. J. S. Tetrahedron Lett. 1992, 33, 6935.
- Aratani, T. Pure Appl. Chem. 1985, 57, 1839.
- Leblanc, Y.; Fitzsimmons, B. J.; Adams, J.; Perez, F.; Rokach, J. J. Org. Chem. 1986, 51, 789.
- Denmark, S. E.; O'Connor, S. P. J. Org. Chem. 1997, 62, 3390.
- Motherwell, W. B.; Roberts, L. R. J. Chem. Soc., Chem. Commun. 1992, 1582.
- De Meijere, A.; Schulz, T. J.; Kostikov, R. R.; Graupner, F.; Murr, T.; Bielfeldt, T. Synthesis 1991, 547.
- Doyle, M. P.; Zhou, Q.-L.; Charnsangavej, C.; Longoria, M. A.; McKervey, M. A.; Garcia, C. F. Tetrahedron Lett. 1996, 37, 4129.