Epigenetics in learning and memory
While the cellular and molecular mechanisms of learning and memory have long been a central focus of neuroscience, it is only in recent years that attention has turned to the epigenetic mechanisms behind the dynamic changes in gene transcription responsible for memory formation and maintenance. Epigenetic gene regulation often involves the physical marking (chemical modification) of DNA or associated proteins to cause or allow long-lasting changes in gene activity. Epigenetic mechanisms such as DNA methylation and histone modifications (methylation, acetylation, and deacetylation) have been shown to play an important role in learning and memory.[1]
DNA Methylation
DNA methylation involves the addition of a methyl group to a 5' cytosine residue. This usually occurs at cytosines that form part of a cytosine-guanine dinucleotide (CpG sites). Methylation can lead to activation or repression of gene transcription and is mediated through the activity of DNA methyltransferases (DNMTs). DNMT3A and DNMT3B regulate de novo methylation of CpG sites, while DNMT1 maintains established methylation patterns.[2] S-adenosyl methionine acts as the methyl donor.[3]
The current hypothesis for how DNA methylation contributes to the storage of memories is that dynamic DNA methylation changes occur temporally to activate transcription of genes that encode for proteins whose role is to stabilize memory.
DNMTs and Memory
Miller and Sweatt demonstrated that rats trained in a contextual fear conditioning paradigm had elevated levels of mRNA for DNMT3a and DNMT3b in the hippocampus.[4] Fear conditioning is an associative memory task where a context, like a room, is paired with an aversive stimulus, like a foot shock; animals who have learned the association show higher levels of freezing behavior when exposed to the context even in the absence of the aversive stimulation. However, when rats were treated with the DNMT inhibitors zebularine or 5-aza-2′-deoxycytidine immediately after fear-conditioning, they demonstrated reduced learning (freezing behavior). When treated rats were re-trained 24 hours later, they performed as well as non-treated rats. Furthermore, it was shown that when these DNMT inhibitors were given 6 hours after training, and the rats were tested 24 hours later, the rats displayed normal fear memory, indicating that DNMTs are involved specifically in memory consolidation.[4] These findings reveal the importance of dynamic changes in methylation status in memory formation.
Feng et al. created double conditional knock out (DKO) mice for the genes DNMT3a and DNMT1. These mice were shown to have significantly weakened long-term potentiation (LTP) and much more easily stimulated long-term depression (LTD) in the hippocampus. When tested in the Morris water navigation task, which is used to study hippocampus-dependent spatial memory, the DNMT3a/DNMT1 DKO mice took longer to find the platform than control mice. Single knock-out mice (SKO) for either DNMT3a or DNMT1 performed normally.[5] DKO mice were also unable to consolidate memory after fear-conditioning. Since SKO mice did not exhibit the same learning and memory defects as the DKO mice, it was concluded that DNMT3a and DNMT1 play redundant roles in regulating learning and memory.
When DNMTs are inhibited in the prefrontal cortex, recall of existing memories is impaired, but not the formation of new ones. This indicates that DNA methylation may be circuit-specific when it comes to regulating the formation and maintenance of memories.[6]
DNA Methylation Targets
The memory suppressor gene, protein phosphatase 1 (PP1), was shown to have increased CpG island methylation after contextual-fear conditioning. This corresponded to decreased levels of PP1 mRNA in the hippocampus of the trained rats. When DNMTs were inhibited, increased methylation at the PP1 gene was no longer observed.[4] These data suggest that during memory consolidation in associative learning tasks, CpG methylation is used to inhibit the expression of PP1, a gene that negatively inhibits memory formation.
Demethylation and Memory
While DNA methylation is necessary to inhibit genes involved in memory suppression, DNA demethylation is important in activating genes whose expression is positively correlated with memory formation. Sweatt and Miller also showed that the gene reelin, which is involved in long term potentiation induction, had a reduced methylation profile and increased reelin mRNA in fear-conditioned versus control rats. Brain-derived neurotrophic factor (BDNF), another important gene in neural plasticity, has also been shown to have reduced methylation and increased transcription in animals that have undergone learning.[7] While these studies have been linked to the hippocampus, recent evidence has also shown increased demethylation of reelin and BDNF in the medial prefrontal cortex (mPFC), an area involved in cognition and emotion.[8]
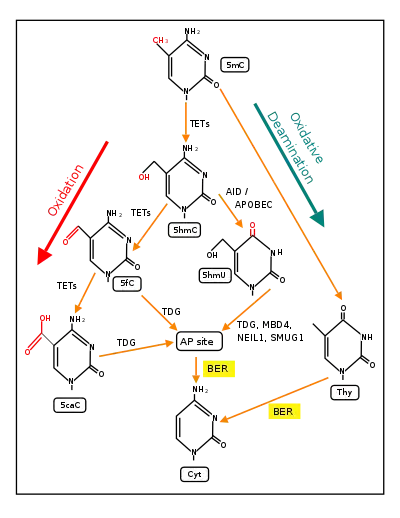
The mechanism behind this experience-dependent demethylation response was previously not fully understood, with some evidence showing that DNMTs may be involved in demethylation.[7] It was also suggested that members of the DNA damage repair GADD45 family may contribute to this demethylation process.[2][3] However, more recently, the pathways illustrated in the Figure below, titled "Demethylation of 5-Methylcytosine (5mC) in neuron DNA," especially the TET dependent pathway, have been confirmed as pathways of DNA demethylation.[9] A role for GADD45 has also recently been indicated, since GADD45 physically interacts with thymine-DNA glycosylase (TDG) and GADD45 may promote the activity of TDG in its role(s) during conversion of 5mC to cytosine.[9]
Methyl-Binding Domain Proteins (MBDs)
Mice that have genetic disruptions for CpG binding protein 2 (MeCP2) have been shown to have significant problems in hippocampus#Role in memory-dependent memory and have impaired hippocampal LTP.[2]
Methylation and Learning and Memory Disorders
Changes in expression of genes associated with post-traumatic stress disorder (PTSD), which is characterized by an impaired extinction of traumatic memory, may be mediated by DNA methylation.[10] In schizophrenics, it has been shown that reelin is down-regulated through increased DNA methylation at promoter regions in GABAergic interneurons. DNMT1 has also been shown to be upregulated in these cells.[10]
Histone methylation
Methylation of histones may either increase or decrease gene transcription depending on which histone is modified, the amino acid that is modified, and the number of methyl groups added.[11] In the case of lysine methylation, three types of modifications exist: monomethylated, dimethylated, or trimethylated lysines. The di- or trimethylation of histone H3 at lysine 9 (H3K9) has been associated with transcriptionally silent regions, while the di- or trimethylation of histone H3 at lysine 4 (H3K4) is associated with transcriptionally active genes.[12]
Histone 3 Lysine 4 Trimethylation and Memory Formation
The hippocampus is an important brain region in memory formation. H3K4 trimethylation is associated with active transcription. In contextual fear conditioning experiments in rats, it was found that levels of H3K4 trimethylation increases in the hippocampus after fear conditioning.[13] In these experiments by Gupta et al., a connection was made between changes in histone methylation and active gene expression during the consolidation of associative memories.[13] In this same study, it was also found that these histone methylations were reversible, as the levels of trimethylation of H3K4 returned to basal levels after a period of 24 hours. This indicated that active demethylation was occurring following memory consolidation. To further explore the role of methyltransferases in long-term memory formation, this study applied the same fear conditioning tests on rats deficient in Mll, a H3K4-specific methyltransferase. The rats with a heterozygous mutant Mll+/- gene showed a significant reduction in their ability to form long-term memories compared to normal rats with an intact Mll gene. Therefore, H3K4 methyltransferases, such as Mll, must have an essential role in long-term memory formation in the hippocampus.[13]
The change in methylation state of histones at the location of specific gene promoters, as opposed to just genome-wide, is also involved in memory formation.[13] Zif268 and BDNF genes are critical for memory consolidation.[14] H3K4 trimethylation increases around both of the Zif268 and BDNF promoters following contextual fear conditioning, when these genes are transcriptionally active. This demonstrates that at the time of memory consolidation, the transcription of memory formation genes such as Zif268 and bdnf is regulated by histone methylation.[13]
Histone 3 Lysine 9 Dimethylation and Memory Formation
Histone H3 lysine 9 dimethylation is associated with transcriptional silencing.[12] The G9a/G9a-like protein (GLP) complex is a methyltransferase specific for producing this modification.[15] One study examined the role of G9a/GLP-mediated transcriptional silencing in the hippocampus and entorhinal cortex (EC) during memory consolidation. It was found that the inhibition of G9a/GLP in the EC, but not in the hippocampus, results in the enhancement of long-term memory formation.[16] In addition, G9a/GLP inhibition in the entorhinal cortex altered histone H3 lysine 9 dimethylation in the Cornu Ammonis area 1 of the hippocampus, suggesting the importance of this complex in mediating connectivity between these two brain regions. Therefore, the G9a/GLP complex plays an important role in histone methylation and long term memory formation in the hippocampus and the EC.[16]
Histone Methylation and Other Epigenetic Modifications
Histone methylation marks are also correlated with other epigenetic modifications, such as histone deacetylation and DNA methylation, in the context of learning and memory. Reduced histone deacetylation is correlated with an increase in H3K9 dimethylation, a modification associated with transcriptional silencing.[13] Therefore, histone deacetylase inhibitors may be applied to increase histone acetylation and suppress H3K9 dimethylation, thereby increasing gene transcription. In the case of DNA methylation, it was found that increases in H3K4 trimethylation correlate with altered DNA methylation of CpG sites at the promoter of Zif268, a gene involved in memory formation, after fear conditioning. Gupta et al. showed that DNA methylation at the Zif268 promoter increased after fear conditioning, correlating with an increase in Zif268 gene expression.[13] This finding was surprising, since it was previously thought that DNA methylation resulted in transcriptional silencing.[13]
Histone Acetylation
Acetylation involves the replacement of a hydrogen with an acetyl group. In a biological context, acetylation is most often associated with the modification of proteins, specifically histones. The acetylation reaction is most often catalyzed by enzymes that contain histone acetyltransferase (HAT) activity.
Histone acetyltransferases (HATs)
HATs are enzymes responsible for the acetylation of amino acids. HATs acetylate by converting the lysine side group of amino acids with the addition of an acetyl group from an acetyl CoA molecule, creating acetyl lysine. HAT enzymes are most often associated with histone proteins and work to regulate the interaction between histones and the DNA that is wrapped around them. HATs are not only restricted to the acetylation of histone but can also acetylate many other proteins implicated in the manipulation of gene expression like that of transcription factors and receptor proteins.
Chromatin Remodeling
Acetylation is one of the main mechanisms implicated in the process of chromatin remodeling. Chromatin remodeling affects the regulation of gene expression by altering the relationship between nucleosomes and DNA. Acetylation of histones removes positive charge, which reduces the level of interaction between the formerly positively charged histone and the negatively charged phosphate groups of the DNA wrapped around the nucleosome complex. This alteration in charges causes a relaxation of DNA from the nucleosome, this relaxed section is seen to have higher levels of gene expression than non acetylated regions.
Acetylation as an epigenetic marker
Patterns of histone acetylation have been useful as a source of epigenetic information due to their ability to reflect changes in transcription rates and the maintenance of gene expression patterns. This acetylation code can then be read and provide generous information for the study of inheritance patterns of epigenetic changes like that of learning, memory and disease states.
Acetlylation as a mechanism for learning and memory
The role of epigenetic mechanisms and chromatin remodeling has been implicated in both synaptic plasticity and neuronal gene expression. Studies with histone deactylase complex inhibitors like SAHA, toluene, garcinol, trichostatin A and sodium butyrate have shown that acetylation is important for the synaptic plasticity of the brain; by inhibiting deactylase complexes total acetylation rates in the brain increased leading to increased rates of transcription and enhanced memory consolidation.[17][18] By using various learning assays like the Morris water maze test and fear conditioning assays in conjunction with acetlyation influencing drugs it was shown that acetylation patterns in the hippocampus are integral to memory association and learning behavior.[19] Studies with various HDAC inhibitors and neural development have shown increased learning and memory, as a result of an increased acetylation state. Conversely studies conducted with HAT inhibitors yielded impairment of memory consolidation and an overall decrease in learning.[20]
ERK/MAPK Cascade
Studies have shown that the ERK/MAPK cascade is important for the regulation of lysine acetylation in the insular cortex of the brain (A part of the brain implicated in the formation of taste memories). The activation of the ERK/MAPK cascade was seen in mice after the introduction of a new taste, the cascade was shown to be necessary for the memory of the taste to be formed. The proposed mechanism for how this cascade works is that MAPK regulates histone acetylation and subsequent chromatin remodeling by means of downstream effectors, such as the CREB binding protein (which has HAT activity).[21][22][23] By observing the rates of acetylation in the insular cortex researchers were able to determine which patterns of acetylation were due to deacetylase or acetylase activity and which were a result of lysine acetyltransferase activity.[22]
Long Term Potentiation
Long term potentiation (LTP) is the enhancement of signal strength between neurons. LTP is the basis of synaptic plasticity and plays a pivotal role in memory formation. LTP is dependent on the activity of NMDA receptors in the brain and it has been shown that NMDA activity influences acetylation. When NMDA receptors are activated they cause an influx of calcium into the cell which in turn activates various signal pathways that ultimately activate the ERK pathway which then modulates transcription factors like CREB. CREB then recruits a HAT to help create and stabilize the long term formation of memory, often through the self-perpetuation of acetylated histones. Studies done on Acetylation of histone H3 in the CA1 region of the hippocampus show that the activation of NMDA receptors increased the acetylation of H3 and conversely inhibition of the ERK pathway in the CA1 region resulted in a decrease in acetylation of H3.[23] In summary:
- NMDA-R activation increases phosphorylation of ERK and Acetylation of Histone H3
- Memory requires proper NMDA-R function
- Memory conditioning increases phosphorylation of ERK and acetylation of Histone H3
- ERK is regulated by phosphorylation
- Histone H3 acetylation is regulated by ERK
- Histone H4 is not regulated by ERK
- HDAC inhibitors enhance LTP, this is dependent on rate of transcription
- HDAC inhibitors do not affect NMDA-R
Histone Deacetylation
HDACs' Role in CREB: CBP-Dependent Transcriptional Activation
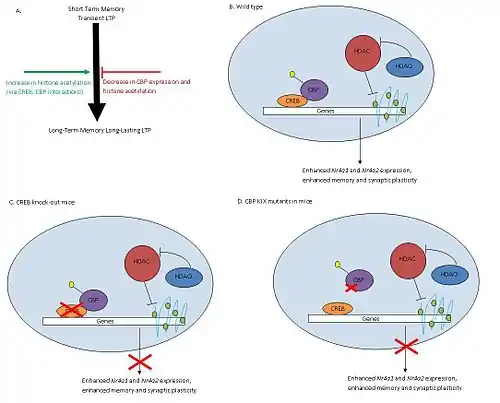
Histone deacetylases (HDAC) remove acetyl groups (-COCH3) from histones altering chromatin structures and decreasing accessibility of transcriptional factors to DNA, thereby reducing transcription of genes. HDACs have shown to play a role in learning and memory through their regulation in the CREB-CBP pathway.
Studies conclude that HDAC inhibitors such as trichostatin A (TSA) increase histone acetylation and improve synaptic plasticity and long-term memory (Fig 1A). CREB, a cAMP response element-binding protein and transcriptional activator, binds CBP forming the CREB: CBP complex. This complex activates genes involved in synaptic formation and long-term memory.(Fig 1B) TSA treatments in the hippocampal CA1 region of mice increased acetylation levels and enhanced long-term potentiation (LTP), a mechanism involved in learning and memory (Fig 1B). However, TSA treatments in CBP mutants lacking KIX domains did not effect LTP in mice (Fig 1D). The KIX domain allows for interaction between CREB and CBP, so knocking out this region disrupts formation of the CREB: CBP complex. Knock outs of CREB produced similar results to those of mutant CBP mice (Fig 1C). Therefore, HDAC inhibition and CREB: CBP association are both necessary for memory development. TSA treatments showed increased expression levels of Nr4a1 and Nra2 genes while other CREB regulated genes were unaffected. HDAC inhibitors improve memory through activation of specific genes regulated by CREB: CBP complex.[24]
HDAC2
The role of individual HDACs in learning and memory is not well understood, but HDAC2 has been shown to negatively regulate memory formation and synaptic plasticity.[19]
Overexpression (OE) of HDAC1 and HDAC2 in mice resulted in decreased levels of acetylated lysines. After exposing these mice to context and tone-dependent fear conditioning experiments, HDAC1 OE mice did not change, but HDAC2 OE mice showed a decrease in freezing behavior, suggesting impairment in memory formation. On the other hand, mice with HDAC2 knockouts (KO) illustrated increased freezing levels compared to wild-type (WT) mice while HDAC1 displayed similar freezing behaviors to WTs. In summary, Guan et al.[19] have shown that:
- HDAC2, not HDAC1, regulates synaptogenesis and synaptic plasticity. HDAC2 overexpression decreases spine density in CA1 pyramidal neurons and dentate gyrus granule cells but HDAC2 KO show an increase in spine density.
- Long term potentiation in CA1 neurons was not observed in HDAC2 OE mice but was easily induced in HDAC2 KO mice. LTP was not altered between HDAC1 KO and OE mice.
- HDAC2 suppresses neuronal gene expression. HDAC2 interacted more than HDAC1 with specific memory-forming promoters such as Bdnf, Egr1, Fos, and GLUR1.
- CoREST, a co-repressor, associates with HDAC2 not HDAC1.
- SAHA, a HDAC inhibitor, increased freezing of HDAC2 OE mice in contextual fear and tone dependent experiments, but did not effect HDAC2 KO mice suggesting HDAC2 is major target of SAHA
HDAC3
HDAC3 is also a negative regulator of long term potentiation formation. McQuown et al.[25] have shown that:
- KOs of HDAC3 in dorsal hippocampus resulted in enhanced memory during object location tests (OLM).
- RGFP136, HDAC3 inhibitor, enhances LTP for object recognition and location
- RGFP136 enhances LTP through CBP-dependent mechanism
- HDAC3 deletions showed increased Nr4a2 and c-Fos expression
- HDAC3 interacts with NCoR and HDAC4 to perform its role in memory formation
HDACs’ Role in CNS disorders
Research has shown that HDACs and HATs play a crucial role in central nervous system (CNS) disorders such as Rett syndrome.[26] Rubinstein-Tabyi syndrome causes mental retardation through possible mutations in CREB-binding protein and p300. However, enhancing expression of CREB-dependent genes or inhibition of HDAC activity partially restore LTP loss and ameliorate late LTP deficits. HDAC inhibitor like TSA may provide a possible therapy for Rubinstein-Tabyi syndrome. Other memory-deficit disorders which may involve HDAC inhibitors as potential therapy are:
Roles of ROS and OGG1 in memory and learning
As reviewed by Massaad and Klann in 2011[29] and by Beckhauser et al. in 2016,[30] reactive oxygen species (ROS) are required for normal learning and memory functions.
One of the most frequent DNA oxidation products of ROS is 8-hydroxy-2'-deoxyguanosine (8-OHdG). Removal of oxidized bases in DNA usually occurs in a matter of minutes, with a half-life of 11 minutes for 8-OHdG.[31] Steady-state levels of endogenous DNA damages represent the balance between formation and repair. 8-OHdGs are among the most frequent DNA damages present in the steady-state, with about 2,400 8-OHdG damaged nucleotides in the average mammalian cell.[32] The steady state 8-OHdG level in the brain is similar to that in other tissues.[33]
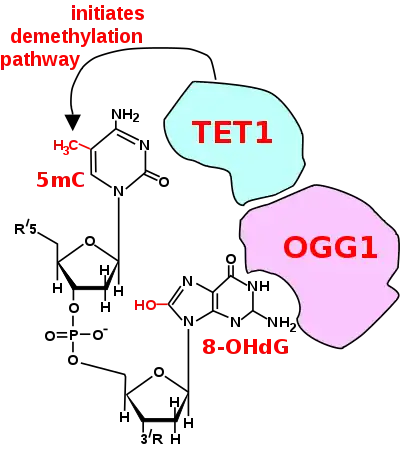
The occurrence of 8-OHdG in neurons appears to have a role in memory and learning. The DNA glycosylase oxoguanine glycosylase (OGG1) is the primary enzyme responsible for the excision of 8-OHdG in base excision repair. However, OGG1, which targets and associates with 8-OHdG, also has a role in adaptive behavior, which implies a physiologically relevant role for 8-OHdG combined with OGG1 in cognition in the adult brain.[34][35] In particular, heterozygous OGG1+/- mice, with about half the protein level of OGG1, exhibit poorer learning performance in the Barnes maze compared to wild-type animals.[36]
In adult somatic cells, such as neurons, DNA methylation typically occurs in the context of CpG dinucleotides (CpG sites), forming 5-methylcytosine (5mC).[27] Thus, a CpG site may be methylated to form 5mCpG. The presence of 5mC at CpG sites in gene promoters is widely considered to be an epigenetic mark that acts to suppress transcription.[37] If the guanine at the 5mCpG site is attacked by ROS, leading to 8-OHdG formation, OGG1 binds to the 8-OHdG lesion without immediate excision of the 8-OHdG. When OGG1 is present at a 5mCp-8-OHdG site, it recruits TET1 to the 8-OHdG lesion and TET1 oxidizes the 5mC adjacent to 8-OHdG. This causes the 5mC to enter the DNA demethylation pathway (see Figure titled "Initiation of DNA demethylation at a CpG site").[27] This pathway is initiated by formation of 5-hydroxymethylcytosine, which may remain in the DNA, or there may be further oxidative reactions followed by base excision repair, to return the nucleoside at that position to cytosine (see Figure "Demethylation of 5-Methylcytosine (5mC) in neuron DNA").
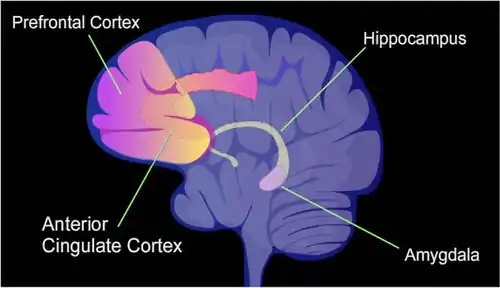
The total number of CpG sites in the human genome is approximately 28 million and the average frequency of CpG sites in the genome is about 1 per hundred base pairs.[38] An intense learning situation can be applied to rats, referred to as contextual fear conditioning.[39] This can result in a life-long fearful memory after a single training event.[39] While the long-term memory of this event appears to be first stored in the hippocampus, this storage is transient and does not remain in the hippocampus.[39] Much of the long term storage of contextual fear conditioning memory appears to take place in the anterior cingulate cortex.[40] (See the Figure showing identified areas of the human brain that are involved in memory formation and also this reference [41].) When contextual fear conditioning is applied to a rat, more than 5,000 differentially methylated regions (DMRs) (of 500 nucleotides each) occur in the rat hippocampus neural genome both one hour and 24 hours after the conditioning in the hippocampus.[42] This causes about 500 genes to be up-regulated (often due to hypomethylation of CpG sites) and about 1,000 genes to be down-regulated (often due to newly formed 5mC at CpG sites in a promoter region). The pattern of induced and repressed genes within neurons appears to provide a molecular basis for forming this first transient memory of this training event in the hippocampus of the rat brain.[42] When similar contextual fear conditioning is applied to a mouse, one hour after contextual fear conditioning there were 675 demethylated genes and 613 hypermethylated genes in the hippocampus region of the mouse brain.[43] These changes were transient in the hippocampal neurons, and almost none were present after four weeks. However, in mice subjected to conditional fear conditioning, after four weeks there were more than 1,000 differentially methylated genes and more than 1,000 differentially expressed genes in the anterior cingulate cortex,[43] where long-term memories are stored in the mouse brain.[40]
References
- Rumbaugh G, Miller CA (2011). "Epigenetic changes in the brain: measuring global histone modifications". Alzheimer's Disease and Frontotemporal Dementia. Methods in Molecular Biology. 670. pp. 263–74. doi:10.1007/978-1-60761-744-0_18. ISBN 978-1-60761-743-3. PMC 3235043. PMID 20967596.
- Bali P, Im HI, Kenny PJ (June 2011). "Methylation, memory and addiction". Epigenetics. 6 (6): 671–4. doi:10.4161/epi.6.6.15905. PMC 3142366. PMID 21586900.
- Lubin FD (2011). "Epigenetic Mechanisms: Critical Contributors to Long-Term Memory Formation". The Neuroscientist. 71 (6): 616–632. doi:10.1177/1073858410386967. PMID 21460188.
- Miller CA, Sweatt JD (March 2007). "Covalent modification of DNA regulates memory formation". Neuron. 53 (6): 857–69. doi:10.1016/j.neuron.2007.02.022. PMID 17359920.
- Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, et al. (April 2010). "Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons". Nature Neuroscience. 13 (4): 423–30. doi:10.1038/nn.2514. PMC 3060772. PMID 20228804.
- Day JJ, Sweatt JD (June 2011). "Epigenetic mechanisms in cognition". Neuron. 70 (5): 813–29. doi:10.1016/j.neuron.2011.05.019. PMC 3118503. PMID 21658577.
- Day JJ, Sweatt JD (November 2010). "DNA methylation and memory formation". Nature Neuroscience. 13 (11): 1319–23. doi:10.1038/nn.2666. PMC 3130618. PMID 20975755.
- Sui L, Wang Y, Ju LH, Chen M (May 2012). "Epigenetic regulation of reelin and brain-derived neurotrophic factor genes in long-term potentiation in rat medial prefrontal cortex". Neurobiology of Learning and Memory. 97 (4): 425–40. doi:10.1016/j.nlm.2012.03.007. PMID 22469747.
- Bayraktar G, Kreutz MR (2018). "The Role of Activity-Dependent DNA Demethylation in the Adult Brain and in Neurological Disorders". Frontiers in Molecular Neuroscience. 11: 169. doi:10.3389/fnmol.2018.00169. PMC 5975432. PMID 29875631.
- Lockett GA, Wilkes F, Maleszka R (October 2010). "Brain plasticity, memory and neurological disorders: an epigenetic perspective". NeuroReport. 21 (14): 909–13. doi:10.1097/wnr.0b013e32833e9288. PMID 20717061.
- Berger SL (May 2007). "The complex language of chromatin regulation during transcription". Nature. 447 (7143): 407–12. Bibcode:2007Natur.447..407B. doi:10.1038/nature05915. PMID 17522673.
- Sims RJ, Nishioka K, Reinberg D (November 2003). "Histone lysine methylation: a signature for chromatin function". Trends in Genetics. 19 (11): 629–39. doi:10.1016/j.tig.2003.09.007. PMID 14585615.
- Gupta S, Kim SY, Artis S, Molfese DL, Schumacher A, Sweatt JD, et al. (March 2010). "Histone methylation regulates memory formation". The Journal of Neuroscience. 30 (10): 3589–99. doi:10.1523/JNEUROSCI.3732-09.2010. PMC 2859898. PMID 20219993.
- Bramham CR (2007). "Control of synaptic consolidation in the dentate gyrus: mechanisms, functions, and therapeutic implications". Progress in Brain Research. 163: 453–71. doi:10.1016/s0079-6123(07)63025-8. ISBN 9780444530158. PMID 17765733.
- Vermeulen M, Mulder KW, Denissov S, Pijnappel WW, van Schaik FM, Varier RA, et al. (October 2007). "Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4". Cell. 131 (1): 58–69. doi:10.1016/j.cell.2007.08.016. PMID 17884155.
- Gupta-Agarwal S, Franklin AV, Deramus T, Wheelock M, Davis RL, McMahon LL, Lubin FD (April 2012). "G9a/GLP histone lysine dimethyltransferase complex activity in the hippocampus and the entorhinal cortex is required for gene activation and silencing during memory consolidation". The Journal of Neuroscience. 32 (16): 5440–53. doi:10.1523/jneurosci.0147-12.2012. PMC 3332335. PMID 22514307.
- Zhao Z, Fan L, Fortress AM, Boulware MI, Frick KM (February 2012). "Hippocampal histone acetylation regulates object recognition and the estradiol-induced enhancement of object recognition". The Journal of Neuroscience. 32 (7): 2344–51. doi:10.1523/jneurosci.5819-11.2012. PMC 3401048. PMID 22396409.
- Huerta-Rivas A, López-Rubalcava C, Sánchez-Serrano SL, Valdez-Tapia M, Lamas M, Cruz SL (July 2012). "Toluene impairs learning and memory, has antinociceptive effects, and modifies histone acetylation in the dentate gyrus of adolescent and adult rats". Pharmacology, Biochemistry, and Behavior. 102 (1): 48–57. doi:10.1016/j.pbb.2012.03.018. PMID 22497993.
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, et al. (May 2009). "HDAC2 negatively regulates memory formation and synaptic plasticity" (PDF). Nature. 459 (7243): 55–60. Bibcode:2009Natur.459...55G. doi:10.1038/nature07925. PMC 3498958. PMID 19424149.
- Stafford JM, Raybuck JD, Ryabinin AE, Lattal KM (July 2012). "Increasing histone acetylation in the hippocampus-infralimbic network enhances fear extinction". Biological Psychiatry. 72 (1): 25–33. doi:10.1016/j.biopsych.2011.12.012. PMC 3352991. PMID 22290116.
- Bousiges O, Vasconcelos AP, Neidl R, Cosquer B, Herbeaux K, Panteleeva I, et al. (December 2010). "Spatial memory consolidation is associated with induction of several lysine-acetyltransferase (histone acetyltransferase) expression levels and H2B/H4 acetylation-dependent transcriptional events in the rat hippocampus". Neuropsychopharmacology. 35 (13): 2521–37. doi:10.1038/npp.2010.117. PMC 3055563. PMID 20811339.
- Swank MW, Sweatt JD (May 2001). "Increased histone acetyltransferase and lysine acetyltransferase activity and biphasic activation of the ERK/RSK cascade in insular cortex during novel taste learning". The Journal of Neuroscience. 21 (10): 3383–91. doi:10.1523/JNEUROSCI.21-10-03383.2001. PMC 6762472. PMID 11331368.
- Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD (September 2004). "Regulation of histone acetylation during memory formation in the hippocampus". The Journal of Biological Chemistry. 279 (39): 40545–59. doi:10.1074/jbc.m402229200. PMID 15273246.
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, et al. (June 2007). "Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation". The Journal of Neuroscience. 27 (23): 6128–40. doi:10.1523/jneurosci.0296-07.2007. PMC 2925045. PMID 17553985.
- McQuown SC, Barrett RM, Matheos DP, Post RJ, Rogge GA, Alenghat T, et al. (January 2011). "HDAC3 is a critical negative regulator of long-term memory formation". The Journal of Neuroscience. 31 (2): 764–74. doi:10.1523/jneurosci.5052-10.2011. PMC 3160172. PMID 21228185.
- Kazantsev AG, Thompson LM (October 2008). "Therapeutic application of histone deacetylase inhibitors for central nervous system disorders". Nature Reviews. Drug Discovery. 7 (10): 854–68. doi:10.1038/nrd2681. PMID 18827828.
- Zhou X, Zhuang Z, Wang W, He L, Wu H, Cao Y, et al. (September 2016). "OGG1 is essential in oxidative stress induced DNA demethylation". Cellular Signalling. 28 (9): 1163–71. doi:10.1016/j.cellsig.2016.05.021. PMID 27251462.
- Bayraktar G, Kreutz MR (2018). "The Role of Activity-Dependent DNA Demethylation in the Adult Brain and in Neurological Disorders". Frontiers in Molecular Neuroscience. 11: 169. doi:10.3389/fnmol.2018.00169. PMC 5975432. PMID 29875631.
- Massaad CA, Klann E (May 2011). "Reactive oxygen species in the regulation of synaptic plasticity and memory". Antioxidants & Redox Signaling. 14 (10): 2013–54. doi:10.1089/ars.2010.3208. PMC 3078504. PMID 20649473.
- Beckhauser TF, Francis-Oliveira J, De Pasquale R (2016). "Reactive Oxygen Species: Physiological and Physiopathological Effects on Synaptic Plasticity". Journal of Experimental Neuroscience. 10 (Suppl 1): 23–48. doi:10.4137/JEN.S39887. PMC 5012454. PMID 27625575.
- Hamilton ML, Guo Z, Fuller CD, Van Remmen H, Ward WF, Austad SN, et al. (May 2001). "A reliable assessment of 8-oxo-2-deoxyguanosine levels in nuclear and mitochondrial DNA using the sodium iodide method to isolate DNA". Nucleic Acids Research. 29 (10): 2117–26. doi:10.1093/nar/29.10.2117. PMC 55450. PMID 11353081.
- Swenberg JA, Lu K, Moeller BC, Gao L, Upton PB, Nakamura J, Starr TB (March 2011). "Endogenous versus exogenous DNA adducts: their role in carcinogenesis, epidemiology, and risk assessment". Toxicological Sciences. 120 Suppl 1: S130-45. doi:10.1093/toxsci/kfq371. PMC 3043087. PMID 21163908.
- Russo MT, De Luca G, Degan P, Parlanti E, Dogliotti E, Barnes DE, et al. (July 2004). "Accumulation of the oxidative base lesion 8-hydroxyguanine in DNA of tumor-prone mice defective in both the Myh and Ogg1 DNA glycosylases". Cancer Research. 64 (13): 4411–4. doi:10.1158/0008-5472.CAN-04-0355. PMID 15231648.
- Marshall P, Bredy TW (2016). "Cognitive neuroepigenetics: the next evolution in our understanding of the molecular mechanisms underlying learning and memory?". NPJ Science of Learning. 1: 16014. Bibcode:2016npjSL...116014M. doi:10.1038/npjscilearn.2016.14. PMC 4977095. PMID 27512601.
- Bjørge MD, Hildrestrand GA, Scheffler K, Suganthan R, Rolseth V, Kuśnierczyk A, et al. (December 2015). "Synergistic Actions of Ogg1 and Mutyh DNA Glycosylases Modulate Anxiety-like Behavior in Mice" (PDF). Cell Reports. 13 (12): 2671–8. doi:10.1016/j.celrep.2015.12.001. PMID 26711335.
- Hofer T, Duale N, Muusse M, Eide DM, Dahl H, Boix F, et al. (May 2018). "Restoration of Cognitive Performance in Mice Carrying a Deficient Allele of 8-Oxoguanine DNA Glycosylase by X-ray Irradiation". Neurotoxicity Research. 33 (4): 824–836. doi:10.1007/s12640-017-9833-7. PMID 29101721.
- Keifer J (February 2017). "Primetime for Learning Genes". Genes. 8 (2): 69. doi:10.3390/genes8020069. PMC 5333058. PMID 28208656.
- Lövkvist C, Dodd IB, Sneppen K, Haerter JO (June 2016). "DNA methylation in human epigenomes depends on local topology of CpG sites". Nucleic Acids Research. 44 (11): 5123–32. doi:10.1093/nar/gkw124. PMC 4914085. PMID 26932361.
- Kim JJ, Jung MW (2006). "Neural circuits and mechanisms involved in Pavlovian fear conditioning: a critical review". Neuroscience and Biobehavioral Reviews. 30 (2): 188–202. doi:10.1016/j.neubiorev.2005.06.005. PMC 4342048. PMID 16120461.
- Frankland PW, Bontempi B, Talton LE, Kaczmarek L, Silva AJ (May 2004). "The involvement of the anterior cingulate cortex in remote contextual fear memory". Science. 304 (5672): 881–3. Bibcode:2004Sci...304..881F. doi:10.1126/science.1094804. PMID 15131309. S2CID 15893863.
- "The Brain - Queensland Brain Institute - University of Queensland".
- Duke CG, Kennedy AJ, Gavin CF, Day JJ, Sweatt JD (July 2017). "Experience-dependent epigenomic reorganization in the hippocampus". Learning & Memory. 24 (7): 278–288. doi:10.1101/lm.045112.117. PMC 5473107. PMID 28620075.
- Halder R, Hennion M, Vidal RO, Shomroni O, Rahman RU, Rajput A, et al. (January 2016). "DNA methylation changes in plasticity genes accompany the formation and maintenance of memory". Nature Neuroscience. 19 (1): 102–10. doi:10.1038/nn.4194. PMC 4700510. PMID 26656643.