Dicobalt octacarbonyl
Dicobalt octacarbonyl is the organometallic compound with composition Co2(CO)8. This metal carbonyl is used as a reagent and catalyst in organometallic chemistry and organic synthesis, and is central to much known organocobalt chemistry.[2][3] It is the precursor to a hydroformylation catalyst, cobalt tetracarbonyl hydride.[4] Each molecule consists of two cobalt atoms bound to eight carbon monoxide ligands, though multiple distinct structural arrangements are known.[5] Some of the carbonyl ligands are highly labile. The compound is highly reactive towards alkynes, and is sometimes used as an alkyne protecting group. As the cobalt-alkyne complex, it plays a role in promoting both the Nicholas reaction[3][6][7] and the Pauson–Khand reaction.[3][8][9]
 | |
 | |
 | |
| Names | |
|---|---|
| IUPAC name
Octacarbonyldicobalt(Co—Co) | |
| Other names
Cobalt carbonyl, di-mu-Carbonylhexacarbonyldicobalt, Cobalt octacarbonyl, Cobalt tetracarbonyl dimer, Dicobalt carbonyl, Octacarbonyldicobalt | |
| Identifiers | |
3D model (JSmol) |
|
| ChemSpider | |
| ECHA InfoCard | 100.030.454 |
| EC Number |
|
PubChem CID |
|
| RTECS number |
|
| UNII | |
| UN number | 3281 |
CompTox Dashboard (EPA) |
|
| |
| |
| Properties | |
| Co2(CO)8 | |
| Molar mass | 341.95 g/mol |
| Appearance | red-orange crystals white crystalline solid when pure[1] |
| Density | 1.87 g/cm3 |
| Melting point | 51 to 52 °C (124 to 126 °F; 324 to 325 K) |
| Boiling point | 52 °C (126 °F; 325 K) c.a. decomposes |
| insoluble | |
| Vapor pressure | 0.7 mmHg (20 °C)[1] |
| Structure | |
| 1.33 D (C2v isomer) 0 D (D3d isomer) | |
| Hazards | |
| Main hazards | Very toxic, evolves CO gas on decomposition,[1] pyrophoric, carcinogenic |
| Safety data sheet | External MSDS |
| GHS pictograms |     |
| GHS Signal word | Danger |
| H228, H251, H302, H312, H315, H317, H319, H330, H334, H350, H351, H361, H412 | |
| NFPA 704 (fire diamond) | |
| Flash point | Pyrophoric [1] |
| NIOSH (US health exposure limits): | |
PEL (Permissible) |
none[1] |
REL (Recommended) |
TWA 0.1 mg/m3[1] |
IDLH (Immediate danger) |
N.D.[1] |
| Related compounds | |
Related metal carbonyls |
Iron pentacarbonyl Diiron nonacarbonyl Nickel tetracarbonyl |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references | |

Synthesis, structure, properties
Dicobalt octacarbonyl is a white solid when of high purity, but more typically is an orange-colored, pyrophoric solid that is thermally unstable.[1] It is synthesised by the high pressure carbonylation of cobalt(II) salts. In the method patented by James Eli Knap, cobalt(II) acetate is heated to between 150 and 200 °C and exposed to hydrogen and carbon monoxide gases at pressures of 2000 to 6000 psi:[10]
- 2 Co(CH3CO2)2 + 8 CO + 2 H2 → Co2(CO)8 + 4 CH3COOH
The preparation is often carried out in the presence of cyanide, converting the cobalt(II) salt into a hexacyanocobaltate(II) complex that is then treated with carbon monoxide to yield K[Co(CO)4]. Acidification produces cobalt tetracarbonyl hydride, HCo(CO)4, which can then be heated to form dicobalt octacarbonyl.[3][11] It can also be prepared by heating cobalt metal to above 250 °C in a stream of carbon monoxide gas at about 200 to 300 atm:[3]
- 2 Co + 8 CO → Co2(CO)8
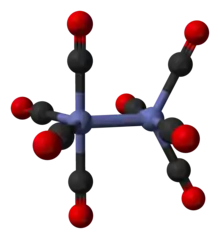
It is known to exist in several isomeric forms, all with the same composition – [Co2(CO)8] – with two cobalt metal centres in oxidation state zero surrounded by eight carbonyl (CO) ligands. These ligands can be bridging between the two cobalt centres or bound to a single metal centre (a terminal ligand).[2][3] In solution, there are two isomers known that rapidly interconvert:[5]
8NoCo-Co.png.webp)
The major isomer (on the left in the above equilibrium process) contains two bridging carbonyl ligands linking the cobalt centres and six terminal carbonyl ligands, three on each metal.[5] It can be summarised by the formula (CO)3Co(μ-CO)2Co(CO)3 and has C2v symmetry. This structure resembles diiron nonacarbonyl (Fe2(CO)9) but with one fewer bridging carbonyl. The Co–Co distance is 2.52 Å, and the Co–COterminal and Co–CObridge distances are 1.80 and 1.90 Å, respectively.[12] Analysis of the bonding suggests the absence of a direct cobalt–cobalt bond.[13]
The minor isomer has no bridging carbonyl ligands, but instead has a direct bond between the cobalt centres and eight terminal carbonyl ligands, four on each metal atom.[5] It can be summarised by the formula (CO)4Co-Co(CO)4 and has D4d symmetry. It features an unbridged cobalt–cobalt bond that is 2.70 Å in length in the solid structure when crystallized together with C60.[14]
Reactions
Nicholas reaction
The Nicholas reaction is a substitution reaction whereby an alkoxy group located on the α-carbon of an alkyne is replaced by another nucleophile.[6][7] The alkyne reacts first with dicobalt octacarbonyl, from which is generated a stabilized propargylic cation[15][16] that reacts with the incoming nucleophile and the product then forms by oxidative demetallation.[6][7]
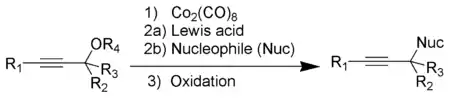
Pauson–Khand reaction
The Pauson–Khand reaction,[8] in which an alkyne, an alkene, and carbon monoxide cyclize to give a cyclopentenone, can be catalyzed by Co2(CO)8,[3][9] though newer methods that are more efficient have since been developed:[17][18]

Co2(CO)8 reacts with alkynes to form a stable covalent complex, which is useful as a protective group for the alkyne. This complex itself can also be used in the Pauson–Khand reaction.[8]
Intramolecular Pauson–Khand reactions, where the starting material contains both the alkene and alkyne moieties, are possible. In the asymmetric synthesis of the Lycopodium alkaloid huperzine-Q, Takayama and co-workers used an intramolecular Pauson–Khand reaction to cyclise an enyne containing a tert-butyldiphenylsilyl (TBDPS) protected primary alcohol.[19] The preparation of the cyclic siloxane moiety immediately prior to the introduction of the dicobalt octacarbonyl ensures that the product is formed with the desired conformation.[20]

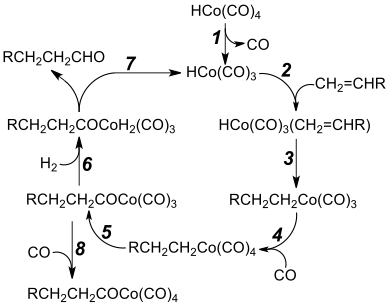
Step 1: Dissociation of carbon monoxide from cobalt tetracarbonyl hydride to form HCo(CO)3, the active catalytic species
Step 2: The cobalt centre forms a π bond to the alkene
Step 3: Alkene ligand inserts into the cobalt–hydride bond
Step 4: Coordination of an additional carbonyl ligand
Step 5: Migratory insertion of a carbonyl ligand into the cobalt–alkyl bond, converting the alkyl tetracarbonyl intermediate into an acyl tricarbonyl species[21]
Step 6: Oxidative addition of dihydrogen leads to a dihydrido complex
Step 7: Aldehyde product released by reductive elimination,[22] regenerating the active catalytic species
Step 8: An unproductive and reversible side reaction
Hydroformylation
Hydrogenation of Co2(CO)8 produces cobalt tetracarbonyl hydride, [HCo(CO)4]:[23]
- Co2(CO)8 + H2 → 2 HCo(CO)4
This hydride is used as a catalyst for hydroformylation – the conversion of a terminal alkene, RCH=CH2, to an aldehyde, RCH2CH2CHO.[4][23] The catalytic cycle for this hydroformylation is shown in the diagram.[4][21][22]
Reduction of Co2(CO)8 with sodium amalgam gives the conjugate base of HCo(CO)4. This salt yields the hydride on acidification, providing an alternative synthetic pathway to that species.[3] Salts of this form are also intermediates in the cyanide synthesis pathway for dicobalt octacarbonyl.[11]
- Co2(CO)8 + 2 Na → 2 NaCo(CO)4
- NaCo(CO)4 + H+ → HCo(CO)4 + Na+
The CO ligands can be replaced with tertiary phosphine ligands to give Co2(CO)8−x(PR3)x. These bulky derivatives are more selective catalysts for hydroformylation reactions.[3] "Hard" Lewis bases, e.g. pyridine, cause disproportionation:
- 12 C5H5N + 3 Co2(CO)8 → 2 [Co(C5H5N)6][Co(CO)4]2 + 8 CO
9.png.webp)
Tricobalt nonacarbonyls
Heating causes decarbonylation and formation of tetracobalt dodecacarbonyl:[3][24]
- 2 Co2(CO)8 → Co4(CO)12 + 4 CO
Like many metal carbonyls, dicobalt octacarbonyl abstracts halides from alkyl halides. Upon reaction with bromoform, it converts to methylidynetricobaltnonacarbonyl, HCCo3(CO)9, by a reaction that can be idealised as:[25]
- 9 Co2(CO)8 + 4 CHBr3 → 4 HCCo3(CO)9 + 36 CO + 6 CoBr2
X-ray crystallographic analysis shows the product contains a triangle of cobalt atoms at distances near 2.48 Å, each bound to three terminal carbonyl groups, and with a methylidyne (CH) group forming the apex of a triangular pyramid. This product is structurally related to tetracobalt dodecacarbonyl,[26] theoretically by replacing the methylidyne group by a fourth Co(CO)3 moiety. However, there has been disagreement between theory and experiment with the actual structure of tetracobalt dodecacarbonyl being shown to have three bridging carbonyl groups.[27][28][29]
Safety
Co2(CO)8 a volatile source of cobalt(0), is pyrophoric and releases carbon monoxide upon decomposition.[30] The National Institute for Occupational Safety and Health has recommended that workers should not be exposed to concentrations greater than 0.1 mg/m3 over an eight-hour time-weighted average, without the proper respiratory gear.[31]
References
- NIOSH Pocket Guide to Chemical Hazards. "#0147". National Institute for Occupational Safety and Health (NIOSH).
- Pauson, Peter L.; Stambuli, James P.; Chou, Teh‐Chang; Hong, Bor‐Cherng (2014). "Octacarbonyldicobalt". Encyclopedia of Reagents for Organic Synthesis. John Wiley & Sons. doi:10.1002/047084289X.ro001.pub3.
- Donaldson, John Dallas; Beyersmann, Detmar (2005). "Cobalt and Cobalt Compounds". Ullmann's Encyclopedia of Industrial Chemistry. Wiley-VCH. doi:10.1002/14356007.a07_281.pub2.
- Elschenbroich, C.; Salzer, A. (1992). Organometallics: A Concise Introduction (2nd ed.). Weinheim: Wiley-VCH. ISBN 3-527-28165-7.
- Sweany, Ray L.; Brown, Theodore L. (1977). "Infrared spectra of matrix-isolated dicobalt octacarbonyl. Evidence for the third isomer". Inorganic Chemistry. 16 (2): 415–421. doi:10.1021/ic50168a037.
- Nicholas, Kenneth M. (1987). "Chemistry and synthetic utility of cobalt-complexed propargyl cations". Acc. Chem. Res. (Review). 20 (6): 207–214. doi:10.1021/ar00138a001.
- Teobald, Barry J. (2002). "The Nicholas reaction: The use of dicobalt hexacarbonyl-stabilised propargylic cations in synthesis". Tetrahedron (Review). 58 (21): 4133–4170. doi:10.1016/S0040-4020(02)00315-0.
- Pauson, P. L.; Khand, I. U. (1977). "Uses of Cobalt-Carbonyl Acetylene Complexes in Organic Synthesis". Ann. N. Y. Acad. Sci. 295 (1): 2–14. doi:10.1111/j.1749-6632.1977.tb41819.x.
- Blanco-Urgoiti, Jaime; Añorbe, Loreto; Pérez-Serrano, Leticia; Domínguez, Gema; Pérez-Castells, Javier (2004). "The Pauson–Khand reaction, a powerful synthetic tool for the synthesis of complex molecules". Chem. Soc. Rev. 33 (1): 32–42. doi:10.1039/b300976a.
- US patent 3236597, Knap, James Eli, "High-Purity Dicobalt Octacarbonyl", issued February 22, 1966, assigned to Union Carbide Corporation
- Orchin, Milton (1953). "Hydrogenation of Organic Compounds with Synthesis Gas". Advances in Catalysis. 5. Academic Press. pp. 385–415. ISBN 9780080565095.
- Sumner, G. Gardner; Klug, Harold P.; Alexander, Leroy E. (1964). "The crystal structure of dicobalt octacarbonyl". Acta Crystallographica. 17 (6): 732–742. doi:10.1107/S0365110X64001803.
- Green, Jennifer C.; Green, Malcolm L. H.; Parkin, Gerard (2012). "The occurrence and representation of three-centre two-electron bonds in covalent inorganic compounds". Chemical Communications. 2012 (94): 11481–11503. doi:10.1039/c2cc35304k.
- Garcia, Thelma Y.; Fettinger, James C.; Olmstead, Marilyn M.; Balch, Alan L. (2009). "Splendid symmetry: Crystallization of an unbridged isomer of Co2(CO)8 in Co2(CO)8·C60". Chemical Communications. 2009 (46): 7143–7145. doi:10.1039/b915083h.
- Lockwood, Rosa F.; Nicholas, Kenneth M. (1977). "Transition metal-stabilized carbenium ions as synthetic intermediates. I. α-[(alkynyl)dicobalt hexacarbonyl] carbenium ions as propargylating agents". Tetrahedron Lett. 18 (48): 4163–4165. doi:10.1016/S0040-4039(01)83455-9.
- Nicholas, K. M.; Pettit, R. (1972). "On the stability of α-(alkynyl)dicobalt hexacarbonyl carbonium ions". J. Organomet. Chem. 44 (1): C21–C24. doi:10.1016/0022-328X(72)80037-8.
- Schore, Neil E. (1991). "The Pauson–Khand Cycloaddition Reaction for Synthesis of Cyclopentenones". Org. React. 40: 1. doi:10.1002/0471264180.or040.01.
- Gibson, Susan E.; Stevenazzi, Andrea (2003). "The Pauson–Khand Reaction: The Catalytic Age Is Here!". Angew. Chem. Int. Ed. 42 (16): 1800–1810. doi:10.1002/anie.200200547.
- Nakayama, Atsushi; Kogure, Noriyuki; Kitajima, Mariko; Takayama, Hiromitsu (2011). "Asymmetric Total Synthesis of a Pentacyclic Lycopodium Alkaloid: Huperzine‐Q". Angew. Chem. Int. Ed. 50 (35): 8025–8028. doi:10.1002/anie.201103550.
- Ho, Tse-Lok (2016). "Dicobalt Octacarbonyl". Fiesers' Reagents for Organic Synthesis. 28. John Wiley & Sons. pp. 251–252. ISBN 9781118942819.
- Heck, Richard F.; Breslow, David S. (1961). "The Reaction of Cobalt Hydrotetracarbonyl with Olefins". Journal of the American Chemical Society. 83 (19): 4023–4027. doi:10.1021/ja01480a017.
- Halpern, Jack (2001). "Organometallic chemistry at the threshold of a new millennium. Retrospect and prospect". Pure and Applied Chemistry. 73 (2): 209–220. doi:10.1351/pac200173020209.
- Pfeffer, M.; Grellier, M. (2007). "Cobalt Organometallics". Comprehensive Organometallic Chemistry III. 7. Elsevier. pp. 1–119. doi:10.1016/B0-08-045047-4/00096-0.
- Chini, P. (1968). "The closed metal carbonyl clusters". Inorganica Chimica Acta Reviews. 2: 31–51. doi:10.1016/0073-8085(68)80013-0.
- Nestle, Mara O.; Hallgren, John E.; Seyferth, Dietmar; Dawson, Peter; Robinson, Brian H. (1980). "μ3-Methylidyne and μ3-Benzylidyne-Tris(Tricarbonylcobalt)". Inorg. Synth. 20: 226–229. doi:10.1002/9780470132517.ch53.
- Leung, P.; Coppens, P.; McMullan, R. K.; Koetzle, T. F. (1981). "The Structure of Nonacarbonyl-μ3-methylidyne-triangulo-tricobalt. X-ray and Neutron Diffraction Studies". Acta Crystallogr. B. 37 (7): 1347–1352. doi:10.1107/S0567740881005906.
- Corradini, Paolo (1959). "Structure of tetracobaltdodecarbonyl". Journal of Chemical Physics. 31 (6): 1676–1677. doi:10.1063/1.1730674.
- Wei, Chin Hsuan (1969). "Structural analyses of tetracobalt dodecacarbonyl and tetrarhodium dodecacarbonyl. Crystallographic treatments of a disordered structure and a twinned composite". Inorganic Chemistry. 8 (11): 2384–2397. doi:10.1021/ic50081a030.
- Farrugia, Louis J.; Braga, Dario; Grepioni, Fabrizia (1999). "A structure redetermination of Co4(CO)12: Evidence for dynamic disorder and the pathway of metal atom migration in the crystalline phase". Journal of Organometallic Chemistry. 573 (1–2): 60–66. doi:10.1016/S0022-328X(98)00879-1.
- Cole Parmer MSDS
- CDC - NIOSH Pocket Guide to Chemical Hazards