Crystallization of polymers
Crystallization of polymers is a process associated with partial alignment of their molecular chains. These chains fold together and form ordered regions called lamellae, which compose larger spheroidal structures named spherulites.[1][2] Polymers can crystallize upon cooling from melting, mechanical stretching or solvent evaporation. Crystallization affects optical, mechanical, thermal and chemical properties of the polymer. The degree of crystallinity is estimated by different analytical methods and it typically ranges between 10 and 80%, with crystallized polymers often called "semi-crystalline". The properties of semi-crystalline polymers are determined not only by the degree of crystallinity, but also by the size and orientation of the molecular chains.
Crystallization mechanisms

Solidification from the melt
Polymers are composed of long molecular chains which form irregular, entangled coils in the melt. Some polymers retain such a disordered structure upon freezing and readily convert into amorphous solids. In other polymers, the chains rearrange upon freezing and form partly ordered regions with a typical size of the order 1 micrometer.[3] Although it would be energetically favorable for the polymer chains to align parallel, such alignment is hindered by the entanglement. Therefore, within the ordered regions, the polymer chains are both aligned and folded. Those regions are therefore neither crystalline nor amorphous and are classified as semicrystalline. Examples of semi-crystalline polymers are linear polyethylene (PE), polyethylene terephthalate (PET), polytetrafluoroethylene (PTFE) or isotactic polypropylene (PP).[4]


Whether or not polymers can crystallize depends on their molecular structure – presence of straight chains with regularly spaced side groups facilitates crystallization. For example, crystallization occurs much easier in isotactic than in the atactic polypropylene form. Atactic polymers crystallize when the side groups are very small, as in polyvinyl and don't crystallize in case of large substituents like in rubber or silicones.[4]
Nucleation

Nucleation starts with small, nanometer-sized areas where as a result of heat motion some chains or their segments occur parallel. Those seeds can either dissociate, if thermal motion destroys the molecular order, or grow further, if the grain size exceeds a certain critical value.[4][5]
Apart from the thermal mechanism, nucleation is strongly affected by impurities, dyes, plasticizers, fillers and other additives in the polymer. This is also referred to as heterogeneous nucleation. This effect is poorly understood and irregular, so that the same additive can promote nucleation in one polymer, but not in another. Many of the good nucleating agents are metal salts of organic acids, which themselves are crystalline at the solidification temperature of the polymer solidification.[5]
Crystal growth from the melt
Crystal growth is achieved by the further addition of folded polymer chain segments and only occurs for temperatures below the melting temperature Tm and above the glass transition temperature Tg. Higher temperatures destroy the molecular arrangement and below the glass transition temperature, the movement of molecular chains is frozen.[6] Nevertheless, secondary crystallization can proceed even below Tg, in the time scale of months and years. This process affects mechanical properties of the polymers and decreases their volume because of a more compact packing of aligned polymer chains.[4][7]
The chains interact via various types of the van der Waals forces. The interaction strength depends on the distance between the parallel chain segments and it determines the mechanical and thermal properties of the polymer.[8]
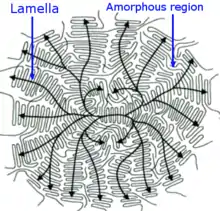
The growth of the crystalline regions preferably occurs in the direction of the largest temperature gradient and is suppressed at the top and bottom of the lamellae by the amorphous folded parts at those surfaces. In the case of a strong gradient, the growth has a unidirectional, dendritic character.[9] However, if temperature distribution is isotropic and static then lamellae grow radially and form larger quasi-spherical aggregates called spherulites. Spherulites have a size between about 1 and 100 micrometers[3] and form a large variety of colored patterns (see, e.g. front images) when observed between crossed polarizers in an optical microscope, which often include the "maltese cross" pattern and other polarization phenomena caused by molecular alignment within the individual lamellae of a spherullite.[2][4][5]
Crystallization by stretching
EN.svg.png.webp)
The above mechanism considered crystallization from the melt, which is important for injection molding of plastic components. Another type of crystallization occurs upon extrusion used in making fibers and films.
In this process, the polymer is forced through, e.g., a nozzle that creates tensile stress which partially aligns its molecules. Such alignment can be considered as crystallization and it affects the material properties. For example, the strength of the fiber is greatly increased in the longitudinal direction, and optical properties show large anisotropy along and perpendicular to the fiber axis. Such anisotropy is more enhanced in presence of rod-like fillers such as carbon nanotubes, compared to spherical fillers.[10] Polymer strength is increased not only by extrusion, but also by blow molding, which is used in the production of plastic tanks and PET bottles.[8] Some polymers which do not crystallize from the melt, can be partially aligned by stretching.[11]
Some elastomers which are amorphous in the unstrained state undergo rapid crystallization upon stretching.
Crystallization from solution
Polymers can also be crystallized from a solution or upon evaporation of a solvent. This process depends on the degree of dilution: in dilute solutions, the molecular chains have no connection with each other and exist as a separate polymer coils in the solution. Increase in concentration which can occur via solvent evaporation, induces interaction between molecular chains and a possible crystallization as in the crystallization from the melt.[12] Crystallization from solution may result in the highest degree of polymer crystallinity. For example, highly linear polyethylene can form platelet-like single crystals with a thickness on the order 10–20 nm when crystallized from a dilute solution. The crystal shape can be more complex for other polymers, including hollow pyramids, spirals and multilayer dendritic structures.[2]
A very different process is precipitation; it uses a solvent which dissolves individual monomers but not the resulting polymer. When a certain degree of polymerization is reached, the polymerized and partially crystallized product precipitates out of the solution. The rate of crystallization can be monitored by a technique which selectively probes the dissolved fraction, such as nuclear magnetic resonance.[12]
Confined crystallization
When polymers crystallize from an isotropic, bulk of melt or concentrated solution, the crystalline lamellae (10 to 20 nm in thickness) are typically organized into a spherulitic morphology as illustrated above. However, when polymer chains are confined in a space with dimensions of a few tens of nanometers, comparable to or smaller than the lamellar crystal thickness or the radius of gyration, nucleation and growth can be dramatically affected. As an example, when a polymer crystallizes in a confined ultrathin layer, the isotropic spherulitic organization of lamellar crystals is hampered and confinement can produce unique lamellar crystal orientations.[13] Sometimes the chain alignment is parallel to the layer plane and the crystals are organized as ‘‘on-edge’’ lamellae. In other cases, "in-plane" lamellae with chain orientation perpendicular to the layers are observed.
The unique crystal orientation of confined polymers imparts anisotropic properties. In one example the large, in-plane polymer crystals reduce the gas permeability of nanolayered films by almost 2 orders of magnitude.[13]
Degree of crystallinity
The fraction of the ordered molecules in polymer is characterized by the degree of crystallinity, which typically ranges between 10% and 80%.[4] Higher values are only achieved in materials having small molecules, which are usually brittle, or in samples stored for long time at temperatures just under the melting point. The latter procedure is costly and is applied only in special cases.
Most methods of evaluating the degree of crystallinity assume a mixture of perfect crystalline and totally disordered areas; the transition areas are expected to amount to several percent. These methods include density measurement, differential scanning calorimetry (DSC), X-ray diffraction (XRD), infrared spectroscopy and nuclear magnetic resonance (NMR). The measured value depends on the method used, which is therefore quoted together with the degree of crystallinity.[4]
In addition to the above integral methods, the distribution of crystalline and amorphous regions can be visualized with microscopic techniques, such as polarized light microscopy and transmission electron microscopy.
| Polymer | D | ρc | ρa |
|---|---|---|---|
| Nylon (PA66 and PA6) | 35–45 | 1.24 | 1.08 |
| Polyoxymethylene (POM) | 70–80 | 1.54 | 1.28 |
| Polyethylene terephthalate (PET) | 30–40 | 1.50 | 1.33 |
| Polybutylene terephthalate (PBT) | 40–50 | – | – |
| Polytetrafluoroethylene (PTFE) | 60–80 | 2.35 | 2.00 |
| isotactic polypropylene | 70–80 | 0.95 | 0.85 |
| atactic polypropylene | ~0 | – | – |
| High-density polyethylene | 70–80 | 1.0 | 0.85 |
| Low-density polyethylene | 45–55 | 1.0 | 0.85 |
- Density measurements
- Crystalline areas are generally more densely packed than amorphous areas. This results in a higher density, up to 15% depending on the material. For example, polyamide 6 (nylon) has crystalline density ρc = 1.24 g/cm3 and amorphous density ρa = 1.08 g/cm3). However, moisture which is often present in the sample does affect this type of measurement.[6]
- Calorimetry
- Additional energy is released upon melting a semicrystalline polymer. This energy can be measured with differential scanning calorimetry and compared with that released upon melting of the standard sample of the same material with known crystallization degree.[6][14]
- X-ray diffraction
- Regular arrangement of atoms and molecules produce sharp diffraction peaks whereas amorphous regions result in broad halos. The diffraction pattern of polymers usually contains a combination of both. Degree of crystallinity can be estimated by integrating the relative intensities of the peaks and halos.[6]
- Infrared spectroscopy (IR)
- Infrared absorption or reflection spectra from crystalline polymers contain additional peaks which are absent in amorphous materials with the same composition. These signals may originate from deformation vibrations of the regular arrangement of molecular chains. From the analysis of these bands, the degree of crystallinity can be estimated.[6]
- Nuclear magnetic resonance (NMR)
- crystalline and amorphous areas differ by the mobility of protons. The latter can be monitored through the line shape of NMR signals and used to estimate the degree of crystallinity.[6]
Kinetics of polymer crystallinity
The methods used to determine the degree of crystallinity can be incorporated over time to measure the kinetics of crystallization. The most basic model for polymer crystallization kinetics comes from Hoffman nucleation theory. The crystallization process of polymers does not always obey simple chemical rate equations. Polymers can crystallize through a variety of different regimes and unlike simple molecules, the polymer crystal lamellae have two very different surfaces. The two most prominent theories in polymer crystallization kinetics are the Avrami equation and Lauritzen-Hoffman Growth Theory.[15]
Properties of semicrystalline polymers
Thermal and mechanical properties
Below their glass transition temperature, amorphous polymers are usually hard and brittle because of the low mobility of their molecules. Increasing the temperature induces molecular motion resulting in the typical rubber-elastic properties. A constant force applied to a polymer at temperatures above Tg results in a viscoelastic deformation, i.e., the polymer begins to creep. Heat resistance is usually given for amorphous polymers just below the glass transition temperature.[16]
Relatively strong intermolecular forces in semicrystalline polymers prevent softening even above the glass transition temperature. Their elastic modulus changes significantly only at high (melting) temperature.[4] It also depends on the degree of crystallinity: higher crystallinity results in a harder and more thermally stable, but also more brittle material, whereas the amorphous regions provide certain elasticity and impact resistance.[2][5] Another characteristic feature of semicrystalline polymers is strong anisotropy of their mechanical properties along the direction of molecular alignment and perpendicular to it.[17]
Above the glass transition temperature amorphous chains in a semi-crystalline polymer are ductile and are able to deform plastically. Crystalline regions of the polymer are linked by the amorphous regions. Tie molecules prevent the amorphous and crystalline phases from separating under an applied load. When a tensile stress is applied the semi-crystalline polymer first deforms elastically. While the crystalline regions remain unaffected by the applied stress, the molecular chains of the amorphous phase stretch. Then yielding, which signifies the onset of plastic deformation of the crystalline regions, occurs.[18]
The molecular mechanism for semi-crystalline yielding involves the deformation of crystalline regions of the material via dislocation motion. Dislocations result in coarse or fine slips in the polymer and lead to crystalline fragmentation and yielding.[19] Fine slip is defined as a small amount of slip occurring on a large number of planes. Conversely, coarse slip is a large amount of slip on few planes. The yield stress is determined by the creation of dislocations and their resistance to motion.[20]
After yielding, a neck is formed in the amorphous region and propagates down the sample length. During necking, the disordered chains align along the tensile direction, forming an ordered structure that demonstrates strengthening due to the molecular reorientation. The flow stress now increases significantly following neck propagation. Mechanical anisotropy increases and the elastic modulus varies along different directions, with a high modulus observed in the draw direction. Drawn semi-crystalline polymers are the strongest polymeric materials due to the stress-induced ordering of the molecular chains.[21]
Other defects, such as voids, occur in the semi-crystalline polymer under tensile stress and can drive the formation of the neck. The voids can be observed via small angle x-ray scattering. Unlike crazes these voids do not transfer stresses.. Notably, cavitation is not observed under compressive stress or shearing. Evidence suggests that cavitation also impacts the onset of yielding. The voids are associated with the breaking of the amorphous phase. The strength of the crystalline phase determines the importance of cavitation in yielding. If the crystalline structures are weak, they deform easily resulting in yielding. Semi-crystalline polymers with strong crystalline regions resist deformation and cavitation, the formation of voids in the amorphous phase, drives yielding.[19]
As done in crystalline materials, particles can be added to semi-crystalline polymers to change the mechanical properties. In crystalline materials the addition of particles works to impede dislocation motion and strengthen the material. However, for many semi-crystalline polymers particle fillers weaken the material. It has been suggested that for particles to have a toughening effect in polymers the interparticle matrix ligament thickness must be smaller than a certain threshold. Crystalline polymers polypropylene and polyethylene display particle strengthening.[22]
Plastics are viscoelastic materials meaning that under applied stress, their deformation increases with time (creep). The elastic properties of plastics are therefore distinguished according to the time scale of the testing to short-time behavior (such as tensile test which lasts minutes), shock loading, the behavior under long-term and static loading, as well as the vibration-induced stress.[23]
Optical properties
Crystalline polymers are usually opaque because of light scattering on the numerous boundaries between the crystalline and amorphous regions. The density of such boundaries is lower and consequentially, the transparency is higher - either for low (amorphous polymer) or high (crystalline) degree of crystallinity.[5] For example, atactic polypropylene is usually amorphous and transparent while syndiotactic polypropylene, which has crystallinity ~50%, is opaque.[24] Crystallinity also affects dyeing of polymers: crystalline polymers are more difficult to stain than amorphous ones because the dye molecules penetrate through amorphous regions with greater ease.[25]
References
- Andrew Keller (1952). "Morphology of crystallizing polymers". Nature. 169 (4309): 913–914. Bibcode:1952Natur.169..913K. doi:10.1038/169913a0. S2CID 4255757.
- Charles E. Carraher; Raymond Benedict Seymour (2003). Seymour/Carraher's polymer chemistry. CRC Press. pp. 43–45. ISBN 978-0-8247-0806-1.
- Linda C. Sawyer; David T. Grubb; Gregory F. Meyers (2008). Polymer microscopy. Springer. p. 5. ISBN 978-0-387-72627-4.
- G. W. Ehrenstein; Richard P. Theriault (2001). Polymeric materials: structure, properties, applications. Hanser Verlag. pp. 67–78. ISBN 978-1-56990-310-0.
- Georg Menges, Edmund Haberstroh, Walter Michaeli, Ernst Schmachtenberg: Plastics Materials Science Hanser Verlag, 2002, ISBN 3-446-21257-4
- GW Becker, Ludwig Bottenbruch, Rudolf Binsack, D. Braun: Engineering Thermoplastics. Polyamides. (in German) Hanser Verlag, 1998 ISBN 3-446-16486-3
- Wilbrand Woebcken, Klaus Stöckhert, HBP Gupta: Plastics Encyclopedia. (in German) Hanser Verlag, 1998, ISBN 3-446-17969-0
- Wolfgang Weissbach: Materials science and materials testing. Vieweg + Teubner Verlag, 2007, ISBN 3-8348-0295-6
- Dendrite in the IWF Knowledge and Media gGmbH (videos and articles on the dendritic crystallization of polypropylene)
- Patil, N; Balzano, L; Portale, G; Rastogi, S (July 2010). "A Study on the Chain−Particle Interaction and Aspect Ratio of Nanoparticles on Structure Development of a Linear Polymer". Macromolecules. 43 (16): 6749–6759. Bibcode:2010MaMol..43.6749P. doi:10.1021/ma100636v. ISSN 0024-9297.
- Michael Thielen, Klaus Hartwig, Peter Gust: Blow molding of plastic articles Hanser Verlag, 2006, ISBN 3-446-22671-0
- J. Lehmann (1966). "The observation of the crystallization of high polymer substances from the solution by nuclear magnetic resonance". Colloid & Polymer Science. 212 (2): 167–168. doi:10.1007/BF01553085. S2CID 96640893.
- Wang, Haopeng; Jong K. Keum; Anne Hiltner; Eric Baer; Benny Freeman; Artur Rozanski; Andrzej Galeski (6 February 2009). "Confined Crystallization of Polyethylene Oxide in Nanolayer Assemblies". Science. 323 (5915): 757–760. Bibcode:2009Sci...323..757W. doi:10.1126/science.1164601. PMID 19197057. S2CID 19276.
- Gottfried W. Ehrenstein, Gabriela Riedel, Pia Trawiel: Practice of thermal analysis of plastics. Hanser Verlag, 2003, ISBN 3-446-22340-1
- Paul C. Painter; Michael M. Coleman (1997). "8". Fundamentals of Polymer Science An Introductory Text, Second Edition. CRC Press.
- Joachim Nentwig: Plastic films (in German) Hanser Verlag, 2006, ISBN 3-446-40390-6
- Martin Bonnet: Plastics in engineering applications: properties, processing and practical use of polymeric materials. (in German) Vieweg+Teubner Verlag, 2008 ISBN 3-8348-0349-9
- Courtney, T. H. "Mechanical Behavior of Materials". Waveland Press (2005) , 392-396
- Pawlak, A., Galeski A,. Rozanski, A. Cavitation during deformation of semicrystalline polymers. Progress in Polymer Science. (2014). 921-958
- Bowden, P.B., Young, R.J. Deformation Mechanisms in Crystalline Polymers. Journal of Materials Science. (1974), 2034-2051.
- Courtney, T. H.. Mechanical Behavior of Materials. Waveland Press (2005) , 392-396
- Bartczak, Z., Argon A.S., Weinberg, M. Toughness mechanism in semi-crystalline polymer blends: II. High-density polyethylene toughened with calcium carbonate filler particles. Polymer, 1999. 2347-2365.
- James F. Shackelford (2009). Introduction to Materials Science for Engineers. Prentice Hall. pp. 168–169. ISBN 978-0-13-601260-3.
- Andrew J. Peacock; Allison R. Calhoun (2006). Polymer chemistry: properties and applications. Hanser Verlag. pp. 286–287. ISBN 978-1-56990-397-1.
- Ágnes Tímár-Balázsy; Dinah Eastop (1998). Chemical principles of textile conservation. Butterworth-Heinemann. p. 11. ISBN 978-0-7506-2620-0.