Bimolecular fluorescence complementation
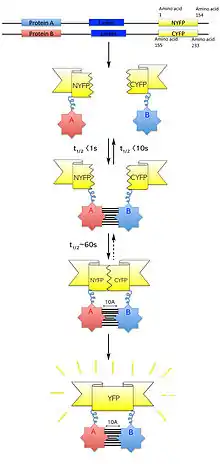
Bimolecular fluorescence complementation (also known as BiFC) is a technology typically used to validate protein interactions. It is based on the association of fluorescent protein fragments that are attached to components of the same macromolecular complex. Proteins that are postulated to interact are fused to unfolded complementary fragments of a fluorescent reporter protein and expressed in live cells. Interaction of these proteins will bring the fluorescent fragments within proximity, allowing the reporter protein to reform in its native three-dimensional structure and emit its fluorescent signal.[1] This fluorescent signal can be detected and located within the cell using an inverted fluorescence microscope that allows imaging of fluorescence in cells. In addition, the intensity of the fluorescence emitted is proportional to the strength of the interaction, with stronger levels of fluorescence indicating close or direct interactions and lower fluorescence levels suggesting interaction within a complex.[2] Therefore, through the visualisation and analysis of the intensity and distribution of fluorescence in these cells, one can identify both the location and interaction partners of proteins of interest.
History
Biochemical complementation was first reported in subtilisin-cleaved bovine pancreatic ribonuclease, then expanded using β-galactosidase mutants that allowed cells to grow on lactose.[3][4][5]
Recognition of many proteins' ability to spontaneously assemble into functional complexes as well as the ability of protein fragments to assemble as a consequence of the spontaneous functional complex assembly of interaction partners to which they are fused was later reported for ubiquitin fragments in yeast protein interactions.[6]
In 2000, Ghosh et al developed a system that allowed a green fluorescent protein (GFP) to be reassembled using an anti-parallel leucine zipper in E. coli cells.[7] This was achieved by dissecting GFP into C- and N-terminal GFP fragments. As the GFP fragment was attached to each leucine zipper by a linker, the heterodimerisation of the anti-parallel leucine zipper resulted in a reconstituted, or re-formed, GFP protein that could be visualised. The successful fluorescent signal indicated that the separate GFP peptide fragments were able to correctly reassemble and achieve tertiary folding. It was, therefore, postulated that using this technique, fragmented GFP could be used to study interaction of protein–protein pairs that have their N–C termini in close proximity.
After the demonstration of successful fluorescent protein fragment reconstitution in mammalian cells, Hu et al. described the use of fragmented yellow fluorescent protein (YFP) in the investigation of bZIP and Rel family transcription factor interactions.[8] This was the first report bZIP protein interaction regulation by regions outside of the bZIP domain, regulation of subnuclear localization of the bZIP domains Fos and Jun by their different interacting partners, and modulation of transcriptional activation of bZIP and Rel proteins through mutual interactions. In addition, this study was the first report of an in vivo technique, now known as the bimolecular fluorescence complementation (BiFC) assay, to provide insight into the structural basis of protein complex formation through detection of fluorescence caused by the assembly of fluorescent reporter protein fragments tethered to interacting proteins.[8]
Fluorescent labeling
Fluorophore activation occurs through an autocatalytic cyclization reaction that occurs after the protein has been folded correctly.[9] This was advanced with the successful reconstitution of the YFP fluorophore from protein fragments that had been fused to interacting proteins within 8 hours of transfection, reported in 2002.[8]
Workflow

Selection of fusion protein production system
There are different production systems that can be used for the fusion protein generated. Transient gene expression is used to identify protein–protein interactions in vivo as well as in subcellular localisation of the BiFC complex. However, one must be cautious against protein over-expression, as this may skew both preferential localisation and the predominant protein complexes formed. Instead, weak promoters, the use of low levels of plasmid DNA in the transfection, and plasmid vectors that do not replicate in mammalian cells should be used to express proteins at or near their endogenous levels to mimic the physiological cellular environment.[10] Also, careful selection of the fluorescent protein is important, as different fluorescent proteins require different cellular environments. For example, GFP can be used in E. coli cells, while YFP is used in mammalian cells.[11]
Stable cell lines with the expression vector integrated into its genome allows more stable gene expression in the cell population, resulting in more consistent results.[1]
Determination of fusion sites
When deciding the linker fusion site on the protein surface, there are three main considerations. First, the fluorescent protein fragments must be able to associate with one another when their tethered proteins interact.[10] Structural information and the location of the interaction surface may be useful when determining the fusion site to the linker, although the information is not necessary, as multiple combinations and permutations can be screened.[10] Secondly, the creation of the fusion protein must not significantly alter the localisation, stability, or expression of the proteins to which the fragments are linked as compared to the endogenous wild-type proteins.[10] Finally, the addition of the fluorescent fragment fusion must not affect the biological function of the protein, preferably verified using assays that evaluate all of the proteins' known functions.[10]
Designing linkers
A linker is a short amino acid sequence that tethers the fluorescent reporter protein fragment to the protein of interest, forming the fusion protein. When designing a linker sequence, one must ensure that the linker is sufficiently soluble and long to provide the fluorescent protein fragments with flexibility and freedom of movement so that the fragment and its partner fragment will collide frequently enough to reconstitute during the interaction of their respective fused proteins.[1] Although it is not documented, it is possible that the length or the sequence of the linker may influence complementation of some proteins.[10] Reported linker sequences RSIAT and RPACKIPNDLKQKVMNH (single amino acid code) and AAANSSIDLISVPVDSR (Sigma) have been successfully used in BiFC experiments.[8][12]
Creating proper plasmid expression vectors
When designing plasmid vectors to express the proteins of interest, the construct must be able to express proteins that are able to form fusion proteins with fluorescent protein fragments without disrupting the protein's function. In addition, the expected protein complex must be able to accept stabilisation of the fluorescent protein fragment interaction without affecting the protein complex function or the cell being studied. Many fluorescent protein fragments that combine in several ways can be used in BiFC.[8][12] Generally, YFP is recommended to serve as the reporter protein, cleaved at residue 155 (N-terminal consisting of residues 1–154 and C-terminal consisting of residues 155–238) or residue 173 in particular, as these sets of fragments are highly efficient in their complementation when fused to many interacting proteins and they produce low levels fluorescence when fused to non-interacting proteins. It is suggested that each target protein is fused to both the N- and C-terminal fragments of the fluorescent reporter protein in turn, and that the fragments are fused at each of the N- and C-terminal ends of the target proteins. This will allow a total of eight different permutations, with interactions being tested:[1]
N-terminal fragment fused at the N-terminal protein 1 + C-terminal fragment fused at the N-terminal protein 2
N-terminal fragment fused at the N-terminal protein 1 + C-terminal fragment fused at the C-terminal protein 2
N-terminal fragment fused at the C-terminal protein 1 + C-terminal fragment fused at the N-terminal protein 2
N-terminal fragment fused at the C-terminal protein 1 + C-terminal fragment fused at the C-terminal protein 2
C-terminal fragment fused at the N-terminal protein 1 + N-terminal fragment fused at the N-terminal protein 2
C-terminal fragment fused at the N-terminal protein 1 + N-terminal fragment fused at the C-terminal protein 2
C-terminal fragment fused at the C-terminal protein 1 + N-terminal fragment fused at the N-terminal protein 2
C-terminal fragment fused at the C-terminal protein 1 + N-terminal fragment fused at the C-terminal protein 2
Selection of appropriate cell culture system
As previously stated, it is important to ensure that the fluorescent reporter protein being used in BiFC is appropriate and can be expressed in the cell culture system of choice, as not all reporter proteins can fluoresce or be visualised in all model systems.
Selection of appropriate controls
Fluorescent protein fragments can associate and fluoresce at low efficiency in the absence of a specific interaction. Therefore, it is important to include controls to ensure that the fluorescence from fluorescent reporter protein reconstitution is not due to unspecific contact.[13]
Some controls include fluorophore fragments linked to non-interacting proteins, as the presence of these fusions tend to decrease non-specific complementation and false positive results.[7]
Another control is created by linking the fluorescent protein fragment to proteins with mutated interaction faces.[8][12] So long as the fluorescent fragment is fused to the mutated proteins in the same manner as the wild-type protein, and the gene expression levels and localisation are unaffected by the mutation, this serves as a strong negative control, as the mutant proteins, and therefore, the fluorescent fragments, should be unable to interact.
Internal controls are also necessary to normalise for differences in transfection efficiencies and gene expression in different cells. This is accomplished by co-transfecting cells with plasmids encoding the fusion proteins of interest as well as a whole (non-fragmented) protein that fluoresces at a different wavelength from the fluorescent reporter protein. During visualisation, one determines the fluorescence intensities of the BiFC complex and the internal control which, after subtracting background signal, becomes a ratio. This ratio represents the BiFC efficiency and can be compared with other ratios to determine the relative efficiencies of the formation of different complexes.[10]
Cell transfection
Once the fusion proteins and controls have been designed and generated in their appropriate expression system, the plasmids must be transfected into the cells to be studied. After transfection, one must wait, typically about eight hours, to allow time for the fusion proteins to interact and their linked fluorescent reporter protein fragments to associate and fluoresce.[8]
Visualisation and analysis
After sufficient time for the fusion proteins and their linked fluorescent fragments to interact and fluoresce, the cells can be observed under an inverted fluorescence microscope that can visualise fluorescence in cells. Although the fluorescence intensity of BiFC complexes is usually <10% of that produced by expression of intact fluorescent proteins, the extremely low autofluorescence in the visible range extremely most cells often makes the BiFC signal orders of magnitude higher than background fluorescence.[14]
If fluorescence is detected when the fusion proteins are expressed, but is lacking or significantly reduced after the expression of the mutated negative control, it is likely that a specific interaction occurs between the two target proteins of interest. However, if the fluorescence intensity is not significantly different between the mutated negative control fusion protein and its wild-type counterpart, then the fluorescence is likely caused by non-specific protein interactions, so a different combination of fusion protein conformations should be tested.
If no fluorescence is detected, an interaction may still exist between the proteins of interest, as the creation of the fusion protein may alter the structure or interaction face of the target protein or the fluorescence fragments may be physically unable to associate. To ensure that this result is not a false negative, that there is no interaction, the protein interaction must be tested in a situation where fluorescence complementation and activation requires an external signal. In this case, if the external signal fails to cause fluorescence fragment association, it is likely that the proteins do not interact or there is a physical impediment to fluorescence complementation.[10]
Strengths
Relevant biological context
Proteins interact with different protein partners and other macromolecules to achieve functions that support different functions in cells that support survival of the organism. Identifying these interactions may provide clues to their effects on cell processes. As these interactions can be affected by both the internal environment and external stimuli, studying these interactions in vivo and at endogenous levels, as is recommended in BiFC, provides a physiologically-relevant context from which to draw conclusions about protein interactions.
Direct visualisation
BiFC enables direct visualisation of protein interactions in living cells with limited cell perturbation, rather than relying on secondary effects or staining by exogenous molecules that can fail to distribute evenly.[1] This, and the ability to observe the living cells for long periods of time, is made possible by the strong intrinsic fluorescence of the reconstituted reporter protein reduces the chances of an incorrect readout associated with the protein isolation process.[1][15]
Sensitivity
Unlike many in vivo protein-interaction assays, BiFC does not require protein complexes to be formed by a large proportion of the proteins or at stoichiometric proportions. Instead, BiFC can detect interactions among protein subpopulations, weak interactions, and low expression proteins due to the stable complementation of the fluorescent reporter protein.[11][16] In addition, successful fluorescent protein reconstitution has been reported for protein partners over 7 nm apart, so long as the linkers binding the fluorophore fragment to the protein of interest has the flexibility needed to associate with its corresponding fragment.[13] Furthermore, the strength of the protein interaction can be quantitatively determined by changes in fluorescent signal strength.[2]
Spatial resolution
BiFC allows measurement of spatial and temporal changes in protein complexes, even in response to activating and inhibiting drugs and subcellularly, providing the highest spatial resolution of in vivo protein–protein interaction assays.[8][17][18]
No specialised equipment
BiFC does not require specialised equipment, as visualisation is possible with an inverted fluorescence microscope that can detect fluorescence in cells.[13] In addition, analysis does not require complex data processing or correction for other sources of fluorescence.[8]
No structural information needed
BiFC can be performed without structural information about the interaction partners, so long as the fluorescent reporter protein fragments can associate within the complex, as multiple combinations of fusion proteins can be screened. This is due to the assumption that, since the protein functions are recapitulated in the in vivo context, the complex structure will resemble that of the intact proteins seen physiologically.[14]
Multiple applications
The BiFC technology has been refined and expanded to include the abilities to simultaneously visualise multiple protein complexes in the same cell, RNA/protein interactions, to quickly detect changes in gene transduction pathways, demonstrate hidden phenotypes of drugs, where the predicted treatment outcome (i.e. cell death, differentiation, morphological change) is not seen in vivo, study complex formation in different cellular compartments, and to map protein interaction surfaces[12][18][19][20][21]
Limitations
Real-time detection
The fluorescent signal only is produced after the proteins have interacted, which is generally in the order of hours. Hence BiFC is unable to provide real-time detection of protein interactions. The delay for chemical reactions to generate fluorophore may also have an effect on the dynamics of complex dissociation and partner exchange.[1][7][8][22]
Irreversible BiFC formation
BiFC complex formation is only reversible during the initial step of fluorescent reporter protein re-assembly, typically in the order of milliseconds. Once the fluorochrome has been reconstituted, it is essentially irreversible in vitro. This prevents proteins from interacting with others and may disrupt the association/disassociation of protein complexes in dynamic equilibrium.[1]
Independent fluorescent protein fragment associations
Fluorescent protein fragments have a limited ability to associate independent of the proteins to which they are fused. Although protein-independent association will vary depending on identities of the fusion proteins and their expression levels, one must provide the necessary and numerous controls to distinguish between true and false-positive protein interactions. Generally, this limitation is mitigated by ensuring that the fusion proteins of interest are expressed at endogenous concentrations.[1]
Altering protein structure and steric hindrance
Fluorescent fragment linkage may alter the folding or structure of the protein of interest, leading to the elimination of an interacting protein's surface binding site. In addition, the arrangement of the fluorescent fragments may prevent fluorophore reconstitution through steric hindrance, although steric hindrance can be reduced or eliminated by using a linker sequence that allows sufficient flexibility for the fluorescent fragments to associate. Therefore, absence of fluorescence complementation may be a false negative and does not necessarily prove that the interaction in question does not occur.
Obligate anaerobes
Due to the requirement of molecular oxygen for fluorophore formation, BiFC cannot be used in obligate anaerobes, which cannot survive in the presence of oxygen. This limits the use of BiFC to aerobic organisms.[1]
Autofluorescence
Autofluorescence is usually not a problem since the BiFC signal will be a lot higher than the background.[23][24] However certain organisms, especially apicomplexa, have a higher autofluorescence that make it harder to apply BiFC in them.[25] Certain fungi, such as Candida albicans, also have a high autofluorescent background, but BiFC can often still be performed when the proper controls and strains are used.[26][27]
Use of fusion proteins
Because endogenous wild-type proteins cannot be visualised in vivo, fusion proteins must be created and their plasmids transfected into the cells studied. These fusion proteins may not recapitulate the functions, localisation, and interactions common to their wild-type counterparts, providing an inaccurate picture of the proteins in question. This problem can be alleviated by using structural information and the location of interaction sites to rationally identify fusion sites on the proteins of interest, using appropriate controls, and comparing the expression levels and functions of the fusion and wild-type proteins through Western Blots and functional assays.[1]
Temperature dependence
Although low temperatures favour the reconstitution of fluorescence when fragments are within proximity, this may affect the behaviour of the target proteins leading to inaccurate conclusions regarding the nature of protein interactions and their interacting partners.[16]
Exact interaction relationship unknown
Because fluorophore reconstitution can occur at a distance of 7 nm or more, fluorescence complementation may indicate either a direct or indirect (i.e. within the same complex) interaction between the fluorescent fragments' fused proteins.[15]
Application
In addition to the validation of protein–protein interactions described above, BiFC has been expanded and adapted to other applications:
Assembly of bacterial ribosomes
The BiFC system has been applied to record ribosome biogenesis events in E.coli.[28] The process of ribosomes assembly involves nucleation of ribosomal proteins in proper order and orientation. Perturbations in assembly can lead to structural defects in ribosomal subunits which as a result cannot join in the correct orientation to form fully functional ribosomes. Thus, the events of subunit joining signaled by the appearance of BiFC is an easy way to monitor ribosome biogenesis in contrast to laborious polysome profiling methods.
Multicolour fluorescence
The fluorescent protein fragments used in BiFC have been expanded to include the colours blue, cyan, green, yellow, red, cherry, and Venus.[8][12][29][30] This range in colours has made the development of multicolour fluorescence complementation analysis possible.[12] This technique allows multiple protein complexes to be visualised simultaneously in the same cell. In addition, proteins typically have a large number of alternative interaction partners. Therefore, by fusing fragments of different fluorescent proteins to candidate proteins, one can study competition between alternative interaction partners for complex formation through the complementation of different fluorescent colour fragments.[12]
RNA-binding protein interactions
BiFC has been expanded to include the study of RNA-binding protein interactions in a method Rackham and Brown described as trimolecular fluorescence complementation (TriFC).[19] In this method, a fragment of the Venus fluorescent protein is fused to the mRNA of interest, and the complementary Venus portion fused to the RNA-binding protein of interest. Similar to BiFC, if the mRNA and protein interact, the Venus protein will be reconstituted and fluoresce. Also known as the RNA bridge method, as the fluorophore and other interacting proteins form a bridge between the protein and the RNA of interest, this allows a simple detection and localisation of RNA-protein interactions within a living cell and provides a simple method to detect direct or indirect RNA-protein association (i.e. within a complex) that can be verified through in vitro analysis of purified compounds or RNAi knockdown of the bridging molecule(s).[19]
Pathway organisation and signal transduction cascades
BiFC can be used to link genes to one other and their function through measurement of interactions among the proteins that the genes encode.[20][21] This application is ideal for novel genes in which little is known about their up- and downstream effectors, as novel pathway linkages can be made. In addition, the effects of drugs, hormones, or deletion or knockdown of the gene of interest, and the subsequent effects on both the strength of the protein–protein interactions and the location of the interaction can be observed within seconds.[17][18]
Complex formation in different cellular compartments
BiFC has been used to study nuclear translocation, via complex localisation, as well as interactions involving integral membrane proteins.[8][31][32][33][34][35][36][37] Thus, BiFC is an important tool in understanding transcription factor localisation in subcellular compartments.
Quantifying protein–protein interaction surfaces
BiFC has been coupled with flow cytometry (BiFC-FC). This allows protein–protein interaction surfaces to be mapped through the introduction of site-directed or random mutations that affect complex formation.[2]
Comparisons to other technologies
Most techniques used to study protein–protein interactions rely on in vitro methods. Unfortunately, studying proteins in an artificial system, outside of their cellular environment, poses a number of difficulties. For example, this may require the removal of proteins from their normal cellular environment. The processing required to isolate the protein may affect its interactions with other proteins. In addition, isolating the protein from the intracellular signaling and mechanisms that occur in the normal cell may provide a misleading picture of intracellular and physiological occurrences.[1] Furthermore, proteins studied in vitro may be studied at concentrations vastly different from their normal abundance levels, may not necessarily be transported efficiently into the cells, or may not be selective enough to function in the host genome.[38][39][40][41] Finally, by studying proteins in vitro, one is unable to determine the influence of specific protein–protein interactions in the cell on the functional or physiological consequences.
Other in vivo assays most commonly used to study protein–protein interactions include fluorescence resonance energy transfer (FRET) and yeast two-hybrid (Y2H) assay. Each of these assays has their advantages and disadvantages in comparison to BiFC:
Fluorescence resonance energy transfer (FRET)
Fluorescence resonance energy transfer (FRET), also known as förster resonance energy transfer, resonance energy transfer (RET) or electronic energy transfer (EET), is based on the transfer of energy from an excited (donor) chromophore or fluorophore (if the chromophores are fluorescent) to a nearby acceptor. In this method, fluorophores are chemically linked or genetically fused to two proteins hypothesised to interact. If the proteins interact, this will bring the fluorophores into close spatial proximity. If the fluorophores are oriented in a manner that exposes the fluorophores to one another, usually ensured when designing and constructing the fluorophore-protein linkage/fusion, then the energy transfer from the excited donor fluorophore will result in a change in the fluorescent intensities or lifetimes of the fluorophores.[1][13]
Yeast two-hybrid (Y2H)
The yeast two-hybrid (Y2H) is a genetic screening technique that can be used to detect physical (binding) protein–protein or protein–DNA interactions. It is usually applied in the model yeast organism Saccharomyces cerevisiae. It tests a 'bait' protein of (un)known function that is fused to, for example, the binding domain of the transcription factor GAL4 against potential interacting proteins or a cDNA library that express , for example, the GAL4 activation domain (the 'prey').[42][43]
Technology comparisons
| Comparison Technology | Similarity to BiFC | Advantages | Disadvantages | |
|---|---|---|---|---|
| FRET | Ability to detect and locate protein interaction sites within live cells[13] | Instantaneous real-time monitoring of protein interactions
Reversible fluorophore interaction |
Close spatial proximity[44]
Decreased sensitivity[44]
Irreversible photo-bleaching[45][46][47]
|
FRET: interaction between protein A and protein B brings the two fluorescent proteins together and energy transfer occurs between the two fluorescent proteins |
| Y2H | In vivo technique used to screen for interactions | Genetic interaction screen
|
Tentative bait-prey linkage[13]
Erroneous transcription activation[13]
Yeast as model organism[48][49]
Overexpression of proteins[15]
Nuclear localisation[15]
|
 Yeast-2-Hybrid: interaction between protein A and protein B activates transcription |
References
- Kerppola, T. K. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat. Protoc. 1, 1278–1286 (2006).
- Morell, M., Espargaro, A., Aviles, F. X. & Ventura, S. Study and selection of in vivo protein interactions by coupling bimolecular fluorescence complementation and flow cytometry. Nat. Protoc. 3, 22–33 (2008).
- Richards, F. M. On the Enzymic Activity of Subtilisin-Modified Ribonuclease. Proc. Natl. Acad. Sci. U. S. A. 44, 162–166 (1958).
- Ullmann, A., Jacob, F. & Monod, J. Characterization by in vitro complementation of a peptide corresponding to an operator-proximal segment of the beta-galactosidase structural gene of Escherichia coli. J. Mol. Biol. 24, 339–343 (1967).
- Ullmann, A., Jacob, F. & Monod, J. On the subunit structure of wild-type versus complemented beta-galactosidase of Escherichia coli. J. Mol. Biol. 32, 1–13 (1968).
- Johnsson, N. & Varshavsky, A. Split ubiquitin as a sensor of protein interactions in vivo. Proc. Natl. Acad. Sci. U. S. A. 91, 10340-10344 (1994).
- Ghosh, I., Hamilton, A. D. & Regan, L. Antiparallel Leucine Zipper-Directed Protein Reassembly: Application to the Green Fluorescent Protein. Journal of the American Chemical Society 122, 5658 (2000).
- Hu, C. D., Chinenov, Y. & Kerppola, T. K. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 9, 789–798 (2002).
- Tsien, R. Y. The green fluorescent protein. Annu. Rev. Biochem. 67, 509–544 (1998).
- "Kerppola Laboratory".
- Kerppola, T. K. Complementary methods for studies of protein interactions in living cells. Nat. Methods 3, 969–971 (2006).
- Hu, C. D. & Kerppola, T. K. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat. Biotechnol. 21, 539–545 (2003).
- Morell, M. et al. Monitoring the interference of protein–protein interactions in vivo by bimolecular fluorescence complementation: the DnaK case. Proteomics 8, 3433–3442 (2008).
- Kerppola, T. K. Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu. Rev. Biophys. 37, 465–487 (2008).
- http://www.vanderbilt.edu/cbi
- Fan, J. Y. et al. Split mCherry as a new red bimolecular fluorescence complementation system for visualizing protein–protein interactions in living cells. Biochem. Biophys. Res. Commun. 367, 47–53 (2008).
- Michnick, S. W., Ear, P. H., Manderson, E. N., Remy, I. & Stefan, E. Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat. Rev. Drug Discov. 6, 569–582 (2007).
- MacDonald, M. L. et al. Identifying off-target effects and hidden phenotypes of drugs in human cells. Nat. Chem. Biol. 2, 329–337 (2006).
- Rackham, O. & Brown, C. M. Visualization of RNA-protein interactions in living cells: FMRP and IMP1 interact on mRNAs. EMBO J. 23, 3346–3355 (2004).
- Remy, I., Wilson, I. A. & Michnick, S. W. Erythropoietin receptor activation by a ligand-induced conformation change. Science 283, 990–993 (1999).
- Remy, I., Montmarquette, A. & Michnick, S. W. PKB/Akt modulates TGF-beta signalling through a direct interaction with Smad3. Nat. Cell Biol. 6, 358–365 (2004)
- Magliery, T. J. et al. Detecting protein–protein interactions with a green fluorescent protein fragment reassembly trap: scope and mechanism. J. Am. Chem. Soc. 127, 146–157 (2005).
- Chen, Hong; Wiedmer, Stefanie; Hanig, Sacha; Entzeroth, Rolf; Kurth, Michael (2013). "Development of Eimeria nieschulzi (Coccidia, Apicomplexa) Gamonts and Oocysts in Primary Fetal Rat Cells". Journal of Parasitology Research. 2013: 591520. doi:10.1155/2013/591520. PMC 3703804. PMID 23862053.
- Kerppola, Tom K (2008). "BIMOLECULAR FLUORESCENCE COMPLEMENTATION (BiFC) ANALYSIS AS A PROBE OF PROTEIN INTERACTIONS IN LIVING CELLS". Annual Review of Biophysics. 37: 465–87. doi:10.1146/annurev.biophys.37.032807.125842. PMC 2829326. PMID 18573091.
- Varea, M; Clavel, A; Doiz, O; Castillo, F.J; Rubio, M.C; Gómez-Lus, R (1 December 1998). "Fuchsin fluorescence and autofluorescence in Cryptosporidium, Isospora and Cyclospora oocysts". International Journal for Parasitology. 28 (12): 1881–1883. doi:10.1016/S0020-7519(98)00146-5. ISSN 0020-7519. PMID 9925267.
- Subotić, Ana; Swinnen, Erwin; Demuyser, Liesbeth; De Keersmaecker, Herlinde; Mizuno, Hideaki; Tournu, Hélène; Van Dijck, Patrick (2017). "A Bimolecular Fluorescence Complementation Tool for Identification of Protein-Protein Interactions in Candida albicans". G3: Genes, Genomes, Genetics. 7 (10): 3509–3520. doi:10.1534/g3.117.300149. PMC 5633398. PMID 28860184.
- Diaz, Giacomo; Polonelli, Luciano; Conti, Stefania; Messana, Irene; Cabras, Tiziana; Putzolu, Martina; Falchi, Angela Maria; Fadda, Maria Elisabetta; Cosentino, Sofia; Isola, Raffaella (2005). "Mitochondrial alterations and autofluorescent conversion of Candida albicans induced by histatins". Microscopy Research and Technique. 66 (5): 219–28. doi:10.1002/jemt.20161. PMID 15940680.
- Sharma, Himanshu; Anand, Baskaran (7 July 2016). "Fluorescence bimolecular complementation enables facile detection of ribosome assembly defects in Escherichia coli". RNA Biology. 13 (9): 872–882. doi:10.1080/15476286.2016.1207037. PMC 5014008. PMID 27388791.
- Jach,G.; Pesch,M.; Richter,K.; Frings,S.; Uhrig,J.F. An improved mRFP1 adds red to bimolecular fluorescence complementation. Nat. Methods. 3, 597–600 (2006)
- Shyu,Y.J., Liu,H., Deng,X., Hu,C.D. Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. BioTechniques. 40, 61–66 (2006).
- de Virgilio, M., Kiosses, W. B. & Shattil, S. J. Proximal, selective, and dynamic interactions between integrin alphaIIbbeta3 and protein tyrosine kinases in living cells. J. Cell Biol. 165, 305–311 (2004).
- Tong, E. H. et al. Regulation of nucleocytoplasmic trafficking of transcription factor OREBP/TonEBP/NFAT5. J. Biol. Chem. 281, 23870-23879 (2006).
- Lopez-Gimenez, J. F., Canals, M., Pediani, J. D. & Milligan, G. The alpha1b-adrenoceptor exists as a higher-order oligomer: effective oligomerization is required for receptor maturation, surface delivery, and function. Mol. Pharmacol. 71, 1015–1029 (2007).
- Nakahara, S., Hogan, V., Inohara, H. & Raz, A. Importin-mediated nuclear translocation of galectin-3. J. Biol. Chem. 281, 39649-39659 (2006).
- Liu, H. et al. Mutual regulation of c-Jun and ATF2 by transcriptional activation and subcellular localization. EMBO J. 25, 1058–1069 (2006).
- Gwozdz, T. et al. EcR and Usp, components of the ecdysteroid nuclear receptor complex, exhibit differential distribution of molecular determinants directing subcellular trafficking. Cell. Signal. 19, 490–503 (2007).
- Fan, M., Ahmed, K. M., Coleman, M. C., Spitz, D. R. & Li, J. J. Nuclear factor-kappaB and manganese superoxide dismutase mediate adaptive radioresistance in low-dose irradiated mouse skin epithelial cells. Cancer Res. 67, 3220–3228 (2007).
- Wu, P., Daniel-Issakani, S., LaMarco, K. & Strulovici, B. An automated high throughput filtration assay: application to polymerase inhibitor identification. Anal. Biochem. 245, 226–230 (1997).
- Stoevesandt, O. & Brock, R. One-step analysis of protein complexes in microliters of cell lysate using indirect immunolabeling & fluorescence cross-correlation spectroscopy. Nat. Protoc. 1, 223–229 (2006).
- Bergendahl, V., Heyduk, T. & Burgess, R. R. Luminescence resonance energy transfer-based high-throughput screening assay for inhibitors of essential protein–protein interactions in bacterial RNA polymerase. Appl. Environ. Microbiol. 69, 1492–1498 (2003).
- Yang, P. et al. Multiplexed detection of protein–peptide interaction and inhibition using capillary electrophoresis. Anal. Chem. 79, 1690–1695 (2007).
- Fields, S. & Song, O. A novel genetic system to detect protein–protein interactions. Nature 340, 245–246 (1989).
- Stynen, B; Tournu, H; Tavernier, J; Van Dijck, P (June 2012). "Diversity in genetic in vivo methods for protein-protein interaction studies: from the yeast two-hybrid system to the mammalian split-luciferase system". Microbiology and Molecular Biology Reviews. 76 (2): 331–82. doi:10.1128/MMBR.05021-11. PMC 3372256. PMID 22688816.
- Kerppola, T. K. Visualization of molecular interactions by fluorescence complementation. Nat. Rev. Mol. Cell Biol. 7, 449–456 (2006).
- Creemers, T. M., Lock, A. J., Subramaniam, V., Jovin, T. M. & Volker, S. Photophysics and optical switching in green fluorescent protein mutants. Proc. Natl. Acad. Sci. U. S. A. 97, 2974–2978 (2000).
- Terskikh, A. et al. "Fluorescent timer": protein that changes color with time. Science 290, 1585–1588 (2000).
- van Thor, J. J., Gensch, T., Hellingwerf, K. J. & Johnson, L. N. Phototransformation of green fluorescent protein with UV and visible light leads to decarboxylation of glutamate 222. Nat. Struct. Biol. 9, 37–41 (2002).
- Stynen, B; Tournu, H; Tavernier, J; Van Dijck, P (June 2012). "Diversity in genetic in vivo methods for protein-protein interaction studies: from the yeast two-hybrid system to the mammalian split-luciferase system". Microbiology and Molecular Biology Reviews. 76 (2): 331–82. doi:10.1128/MMBR.05021-11. PMC 3372256. PMID 22688816.
- Schoeters, F; Van Dijck, P (2019). "Protein-Protein Interactions in Candida albicans". Frontiers in Microbiology. 10: 1792. doi:10.3389/fmicb.2019.01792. PMC 6693483. PMID 31440220.